Sickle cell anemia
Reading File
Finding Sources
Searching PubMed
"sickle cell anemia"[MeSH Terms] AND treatment
Finding Sources
Searching PubMed
"sickle cell disease" AND hydroxyurea
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading Article
PMID: 41831848
I now have all the information needed for a comprehensive response. Let me compile the full answer.
Sickle Cell Anemia
Definition & Genetics
Sickle cell anemia (HbSS) is the most common familial hemolytic anemia and the prototypic hemoglobinopathy. It results from a single point mutation in the β-globin gene on chromosome 11: an adenine-to-thymidine transversion at codon 6 (GAG→GTG), substituting valine for glutamic acid at position 6 of the β-globin chain. This creates sickle hemoglobin (HbS).
- Homozygous HbSS = sickle cell anemia (most severe)
- HbSC, HbSβ+ thalassemia = milder variants
- HbSβ0 thalassemia = severity similar to HbSS
- HbAS (sickle trait) = heterozygous carrier; generally asymptomatic
The HbS allele is prevalent in sub-Saharan Africa, parts of India, southern Europe, and the Middle East because heterozygous carriers have ~90% protection against severe falciparum malaria. In the United States, ~8% of African Americans are HbS carriers; ~1 in 600 have sickle cell anemia. Globally, 300,000-400,000 births with HbSS occur annually, >75% in sub-Saharan Africa.
Pathogenesis
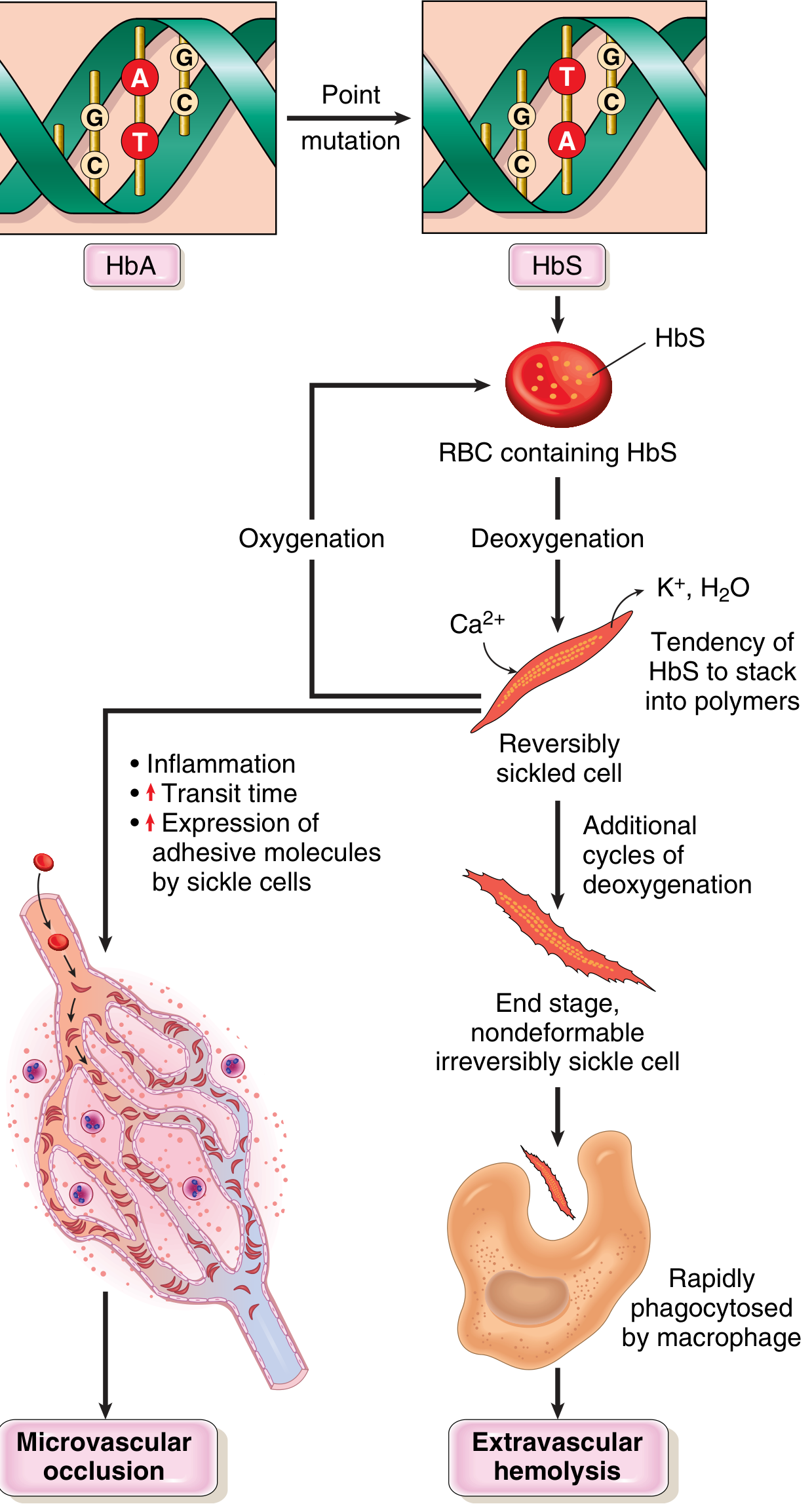
The entire disease flows from one molecular event: deoxygenated HbS polymerizes.
-
HbS polymerization: On deoxygenation, the abnormal valine residue allows intermolecular contacts that form rigid polymers. These distort the red cell into an elongated, crescentic "sickle" shape.
-
Reversible vs. irreversible sickling: Early sickling is reversible on re-oxygenation. However, repeated sickling cycles cause calcium influx, loss of potassium and water, and membrane skeleton damage - eventually creating irreversibly sickled cells (ISCs) prone to hemolysis.
-
Three key factors that determine whether clinically significant sickling occurs:
- Intracellular HbS concentration - HbA and HbF retard polymerization; HbF explains why neonates are protected until ~5-6 months of age; α-thalassemia coexistence reduces Hb concentration and decreases sickling
- Red cell dehydration - increases intracellular Hb concentration, promoting polymerization
- Microvascular transit time - slow blood flow (spleen, bone marrow, inflamed tissues) allows polymerization to occur before cells re-oxygenate
-
Vaso-occlusion mechanism: Beyond simple physical blockage, sickle cells adhere abnormally to endothelium via P-selectin and other adhesion molecules. Leukocytes and platelets are co-recruited. Intravascular hemolysis releases free hemoglobin that scavenges nitric oxide (NO), impairing vasodilation and promoting a pro-thrombotic, pro-inflammatory vascular state.
-
Two major pathologic consequences:
- Hemolytic anemia - RBC lifespan only ~20 days (vs. normal 120 days)
- Vaso-occlusive ischemia - causes pain crises and end-organ damage
Peripheral Blood Smear
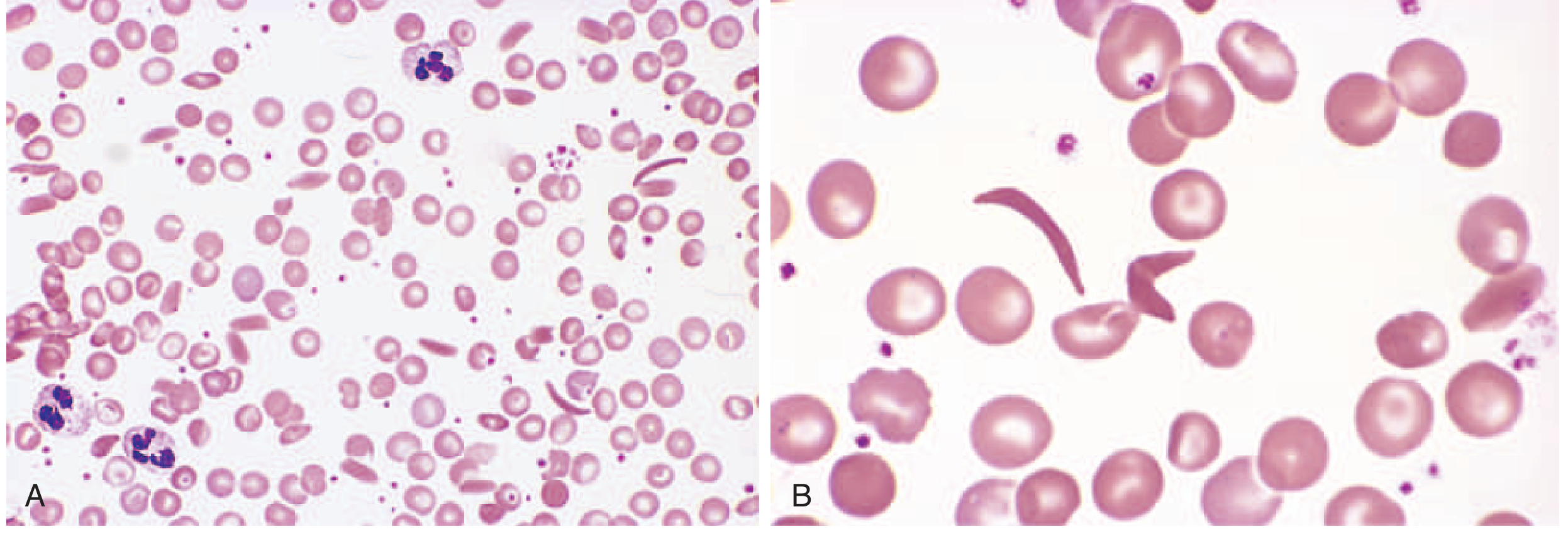
Panel A (low power) and Panel B (high power) show the characteristic elongated, crescent/boat-shaped sickle cells alongside target cells and other poikilocytes.
Morphology & Organ Pathology
| Organ/System | Pathology |
|---|---|
| Spleen | Childhood: moderate splenomegaly (red pulp congestion). Adulthood: autosplenectomy (fibrotic nubbin) from repeated infarcts |
| Bone marrow | Compensatory erythroid hyperplasia → bone resorption → frontal bossing, "crew-cut" skull on X-ray |
| Liver/Heart/Kidney | Hypoxia-induced fatty change |
| Bone | Avascular necrosis (femoral/humeral heads), osteomyelitis |
| Brain | Stroke (ischemic) |
| Lung | Acute chest syndrome |
| Retina | Proliferative retinopathy → blindness |
| Penis | Priapism → penile fibrosis |
Clinical Features
Patients are asymptomatic until ~6 months of age (when HbF switches to HbS). Hematocrit is typically 18-30%.
Vaso-occlusive Crises
- Pain crisis (most common) - precipitated by infection, dehydration, cold, hypoxia, acidosis
- Hand-foot syndrome (dactylitis) - most common presenting symptom in young children; infarction of metacarpal/metatarsal bones
- Acute chest syndrome (ACS) - fever, chest pain, hypoxemia, pulmonary infiltrates; triggered by infection or fat emboli from infarcted bone; vicious cycle of worsening pulmonary and systemic hypoxemia
- Stroke - both ischemic and hemorrhagic; the two leading causes of ischemia-related death are ACS and stroke
- Aplastic crisis - sudden drop in red cell production, typically from parvovirus B19 infection of erythroblasts; self-limited
- Splenic sequestration crisis (mainly in young children before autosplenectomy) - massive pooling of blood in spleen → rapid anemia + hypovolemia
Chronic Complications
- Chronic kidney disease / sickle cell nephropathy
- Pulmonary hypertension
- Leg ulcers
- Gallstones (pigmented; from chronic hemolysis)
- Avascular necrosis
- Retinopathy
Infections
Functionally asplenic patients (both children and adults) are highly susceptible to encapsulated bacteria (especially Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis) and gram-negatives (Salmonella, E. coli - particularly associated with osteomyelitis).
Diagnosis
- Newborn screening: Mandatory in the US - hemoglobin electrophoresis/HPLC from heel-stick blood
- Peripheral smear: Sickle cells, target cells, Howell-Jolly bodies (post-autosplenectomy)
- Hemoglobin electrophoresis / HPLC: Gold standard - quantifies HbS, HbA, HbF, HbA2
- Sickle preparation / Sickledex: Rapid screening; positive in all sickle hemoglobinopathies (not specific for HbSS; false-negatives in neonates with high HbF)
- Prenatal diagnosis: Fetal DNA from amniocentesis or chorionic villus sampling
Lab findings: Normocytic/normochromic anemia, elevated reticulocytes, elevated bilirubin (indirect), elevated LDH, low haptoglobin
Genotype-Severity Table
| Genotype | Hematocrit | HbS% | Severity |
|---|---|---|---|
| HbSS | 18-28% | >90% | Most severe |
| HbSβ0 thalassemia | 18-28% | >90% | Most severe |
| HbSC | 25-35% | ~50% | Moderate |
| HbSβ+ thalassemia | 25-40% | 55-75% | Mild-moderate |
| HbAS (trait) | Normal | 35-40% | Asymptomatic |
Treatment
Disease-Modifying Therapies
| Agent | Mechanism | Key Points |
|---|---|---|
| Hydroxyurea | Increases HbF synthesis (by inducing γ-globin); also reduces WBC count and endothelial adhesion | First-line disease-modifying therapy; reduces pain crises, ACS, hospitalizations, and mortality; NOT used in pregnancy |
| L-glutamine | Reduces oxidative stress in sickle RBCs | FDA-approved; reduces painful crises |
| Voxelotor | Increases hemoglobin oxygen affinity → reduces sickling | Increases Hb levels; reduces hemolysis |
| Crizanlizumab | Anti-P-selectin monoclonal antibody | Reduces vaso-occlusive crises by blocking cell adhesion |
Curative Options
- Hematopoietic stem cell transplantation (HSCT): Potentially curative; best outcomes with HLA-matched sibling donor; significant morbidity/mortality risk
- Gene therapy: Now approved for clinical use (as of 2023-2025) - includes lentiviral gene addition (betibeglogene, lovotibeglogene) and CRISPR-based gene editing (exa-cel/Casgevy); represents a major advance
Transfusion Therapy
- Simple transfusion: For symptomatic anemia, aplastic crisis, splenic sequestration
- Exchange transfusion (RBC exchange): Indicated for stroke, rapidly progressive ACS, multi-organ failure, priapism unresponsive to other treatment, and pre-operative preparation. Goal: HbS <30-40%, Hct <30% (to avoid hyperviscosity)
- Chronic transfusion: Children with prior stroke or transcranial Doppler (TCD) velocity >200 cm/sec
Acute Pain Crisis Management
- Aggressive hydration (IV or oral)
- Analgesia - NSAIDs + opioids (escalate as needed); patient-controlled analgesia (PCA) for severe crises
- Supplemental oxygen (if hypoxic)
- Incentive spirometry (reduces pulmonary complications)
- Treat precipitating factors (infection, dehydration)
Preventive Care
- Penicillin prophylaxis: From 2 months of age through at least age 5 (prevents pneumococcal sepsis)
- Vaccinations: Pneumococcal (PCV + PPSV23), meningococcal, Hib, influenza, hepatitis B
- Folic acid supplementation: Daily, to support erythropoiesis
- TCD screening: Annual in children 2-16 years to identify stroke risk
- Ophthalmology: Annual retinal exams from adolescence
- Hydroxyurea: Start consideration from infancy (≥9 months per current guidelines)
Pregnancy
- Hydroxyurea is contraindicated (teratogenic)
- NSAIDs avoided after 30 weeks
- Regional anesthesia preferred over general
- Prophylactic transfusions considered to reduce pain crises and maternal mortality
- Sixfold increased risk of maternal death compared to unaffected controls
Prognosis
The clinical course is highly variable. Median life expectancy in high-income countries is now into the 5th decade with optimal care. Leading causes of death include ACS, stroke, infection, and multi-organ failure. Gene therapy represents the first realistic path to a cure at population scale.
Sources:
- Robbins & Kumar Basic Pathology (Robbins Pathology), pp. 386-388
- Goldman-Cecil Medicine International Edition (Cecil), pp. 1717-1722
- Harriet Lane Handbook, 23rd ed., p. 479
- Colombatti R et al., Sickle cell disease. Lancet 2026 - comprehensive 2026 update confirming gene therapies now approved and ongoing optimization of hydroxyurea protocols [PMID: 41831848]
This is a shared conversation. Sign in to Orris to start your own chat.