I now have everything needed from the textbooks. Let me build a comprehensive, logically flowing PNH master note structured the way you described — cause → pathogenesis → clinical → labs → smear — designed specifically for active recall retention.
Paroxysmal Nocturnal Hemoglobinuria (PNH) — Master Flow
Sources: Robbins & Kumar Pathologic Basis of Disease, Harrison's Principles of Internal Medicine 22E
The ONE anchor concept (build everything from this)
PNH = acquired somatic mutation in PIGA → no GPI anchors → no CD55/CD59 on blood cells → complement runs wild
Everything else is a consequence of that single sentence.
1. Genetics & Cause
PIGA gene (Phosphatidylinositol Glycan complementation group A):
- X-linked gene
- Encodes an enzyme that catalyzes the first step in GPI anchor biosynthesis
- Because it's X-linked, a single somatic mutation in the active PIGA allele is enough — you don't need two hits (lyonization = random X-inactivation in females means one active copy, one hit knocks it out)
- The mutation occurs in a hematopoietic stem cell (HSC) → all clonal progeny (RBCs, WBCs, platelets) are GPI-deficient
Harrison's adds: In rare cases, biallelic mutations in PIGT (chromosome 20q, autosomal) can cause PNH without a PIGA mutation — because PIGT encodes another enzyme in the same GPI biosynthetic pathway.
Why does the mutant clone expand?
- Normal individuals actually do harbor small numbers of PIGA-mutant marrow cells — they don't cause disease because they have no selective advantage
- In PNH, the clone expands when autoimmune attack targets GPI-linked antigens on normal HSCs → the PIGA-mutant cells (lacking GPI antigens) are spared and gain a growth advantage
- This explains the strong association with aplastic anemia (autoimmune marrow failure)
2. Pathogenesis
GPI anchor normally tethers complement inhibitors to the cell surface:
| GPI-linked Protein | Function |
|---|
| CD55 (DAF — Decay Accelerating Factor) | Inhibits C3 convertase → prevents amplification of complement |
| CD59 (MIRL — Membrane Inhibitor of Reactive Lysis) | Blocks formation of C5b-9 MAC; most important one |
| C8-binding protein | Also inhibits MAC |
Without these → complement goes unchecked:
- Alternative complement pathway activates spontaneously → no brakes
- C3 fragments (C3d) coat the RBC surface
- C5b-9 Membrane Attack Complex (MAC) punches holes in the RBC → intravascular hemolysis
- NO (nitric oxide) released by hemolyzed RBCs is scavenged by free hemoglobin → endothelial dysfunction → thrombosis
Why "nocturnal"?
- During sleep, blood pH drops slightly (mild respiratory acidosis from hypoventilation/CO₂ accumulation) → mildly acidic environment → activates the alternative complement pathway
- BUT: only 25% of cases actually have dramatic paroxysmal nocturnal hemoglobinuria — chronic, constant low-grade hemolysis is the more typical picture
3. Clinical Features
| Feature | Mechanism |
|---|
| Hemoglobinuria (dark/cola-colored urine, especially in the morning) | Intravascular hemolysis → free Hb filtered by kidneys |
| Chronic hemolytic anemia | Ongoing complement-mediated lysis |
| Iron deficiency | Urinary iron loss (hemosiderinuria) — heme iron lost in urine over time |
| Venous thrombosis ← #1 cause of death | NO scavenging by free Hb → platelet activation; MAC-mediated endothelial damage; ~40% of patients. Sites: hepatic vein (Budd-Chiari), portal vein, cerebral veins |
| Aplastic anemia association | Same autoimmune process that destroys normal HSCs; PNH and aplastic anemia coexist |
| Myeloid malignancy (~5%) | AML or myelodysplastic neoplasm — reflects background genetic instability in the HSC |
4. Lab Investigations
| Test | Finding | Why |
|---|
| Flow cytometry (gold standard) | Absent/reduced CD55 & CD59 on RBCs and granulocytes | Direct detection of GPI-deficient cells |
| FLAER (fluorescent aerolysin) | Binds GPI anchors — absent binding in PNH cells | More sensitive than CD55/CD59 alone |
| CBC | Normocytic or microcytic anemia (if iron-deficient) | Hemolysis + iron loss |
| LDH | Elevated | Marker of intravascular hemolysis |
| Haptoglobin | Low/undetectable | Free Hb saturates haptoglobin |
| Hemoglobinuria | Positive urine dipstick for blood (no RBCs on microscopy) | Free Hb passes through glomerulus |
| Hemosiderinuria | Hemosiderin in urine sediment | Chronic iron deposition in renal tubular cells, then sloughed |
| Coombs test (DAT) | Negative | No antibodies — pure complement-mediated, not immune |
| Reticulocyte count | Elevated | Bone marrow compensation |
| Sucrose lysis / Ham's test | Positive (historical, now replaced by flow) | Complement activation in acidic/sucrose environment lyses PNH cells |
| Serum iron, ferritin | Low (iron deficiency from urinary loss) | Hemosiderinuria |
The classic old tests (Ham's acid lysis test, sucrose lysis test) — acidic/low ionic strength → activates complement → PNH cells lyse. Now replaced by flow cytometry.
5. Peripheral Blood Smear
- No specific finding that diagnoses PNH (it's an intravascular hemolytic anemia without RBC structural defects)
- No spherocytes (not antibody-mediated)
- No schistocytes (not a microangiopathic process)
- May see polychromasia (reticulocytosis from compensatory erythropoiesis)
- If iron-deficient: microcytes, pencil cells, hypochromic RBCs
- Pancytopenia on CBC if there's associated aplastic anemia
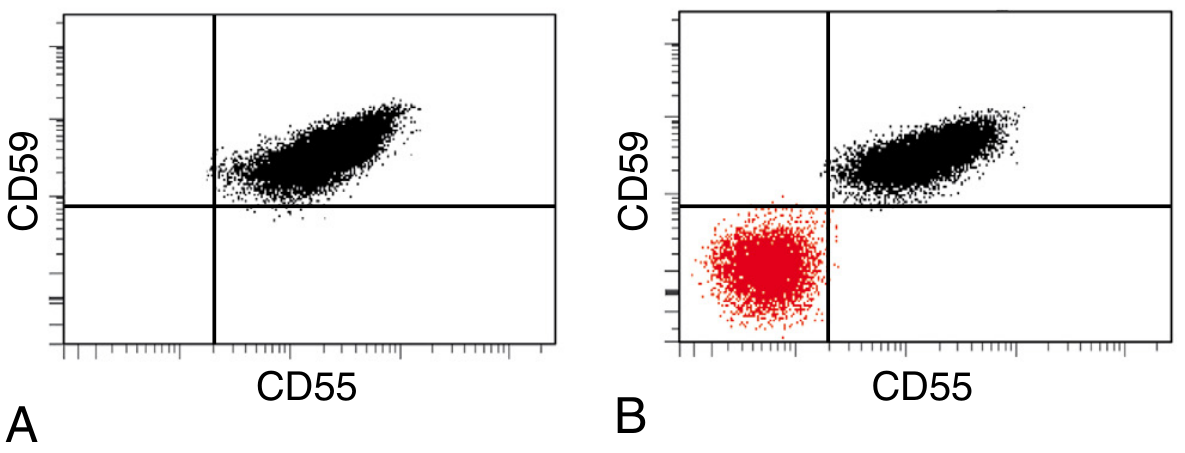
6. Flow Cytometry (Gold Standard)
(A) Normal: All RBCs cluster in the upper-right quadrant — both CD55⁺ and CD59⁺
(B) PNH: A large red population in the lower-left quadrant — CD55⁻ CD59⁻ (the PNH clone). A second normal population remains (from residual normal HSCs).
7. Treatment
| Option | Notes |
|---|
| Eculizumab (anti-C5 monoclonal Ab) | Blocks C5 cleavage → prevents MAC formation → stops intravascular hemolysis. Given IV every 14 days. Side effect: meningococcal infection risk (vaccinate first). Limitation: C3-opsonized PNH cells still undergo extravascular hemolysis (Coombs turns positive) |
| Ravulizumab | Longer-acting anti-C5 (every 8 weeks) |
| Pegcetacoplan (anti-C3) | Upstream C3 inhibition → blocks both intra- and extravascular hemolysis (corrects anemia more completely) |
| Iron/folate supplementation | Replace losses |
| Anticoagulation | For thrombotic episodes |
| Hematopoietic stem cell transplant | Only cure; reserved for aplastic anemia or myeloid malignancy co-occurrence |
The 60-Second Active Recall Chain
PIGA mutated (X-linked, somatic, in HSC)
→ No GPI anchor synthesis
→ No CD55, no CD59, no C8-BP on cell surface
→ Alternative complement runs freely
→ C5b-9 MAC forms on RBC
→ Intravascular hemolysis
→ Hemoglobinuria (morning, dark urine)
→ Free Hb scavenges NO → thrombosis (hepatic/portal/cerebral veins)
→ Urinary iron loss → iron deficiency
Night → CO₂ retention → mild pH drop → complement activated more → paroxysms
Diagnosis: Flow cytometry (CD55/CD59 absent on RBC + granulocytes)
Smear: Non-specific (polychromasia ± microcytes if iron deficient)
Coombs: NEGATIVE
Treatment: Eculizumab (anti-C5) → cure = HSCT
Why You're Forgetting (and what to do about it)
What happened on day 6 is textbook Ebbinghaus forgetting curve — without retrieval, memory decays exponentially. The fact that you retained 98% on day 1 and 50%+ on day 3 means your encoding was excellent. The drop to ~10% by day 6 without another review is completely expected.
The fix: spaced repetition with active recall before re-reading. Before looking at notes, close your eyes and try to reconstruct the chain above (takes 2 minutes). Then check what you missed. That retrieval attempt — even a failed one — is what builds durable memory. The next review should be around day 14, then day 30.
The chain at the end of this note is specifically designed for that: one connected logical thread so you can regenerate the entire topic from a single anchor: "PIGA → no GPI → no CD55/CD59 → complement → hemolysis/thrombosis."
— Robbins & Kumar Pathologic Basis of Disease, p. 606 | Harrison's Principles of Internal Medicine 22E, Ch. 105