I now have everything needed. Here is the complete, structured PNH recap — built as a flow you can mentally walk through from first principles:
Paroxysmal Nocturnal Hemoglobinuria (PNH) — Complete Flow
1. CAUSE (Etiology)
Acquired somatic mutation in the PIGA gene (also written PIG-A) on the short arm of the X chromosome, occurring in a pluripotent hematopoietic stem cell.
Key points to lock in:
- It is the only hemolytic anemia caused by an acquired genetic defect (all others are congenital or immune-mediated)
- X-linked gene → a single mutation is sufficient (no need for a second hit) because the other X is already inactivated (lyonization in females; only one X in males)
- The mutation hits a stem cell, so all progeny — RBCs, WBCs, platelets — carry the defect
- Normal individuals also carry rare PIGA-mutant cells (~1 in 50,000 RBCs), but they don't expand. In PNH, a second step (likely autoimmune attack on normal GPI+ cells) gives the mutant clone a selective advantage
- This explains the strong association with aplastic anemia (AA) — autoimmune marrow damage selects for the GPI-deficient clone that "hides" from immune attack
— Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 606; Goldman-Cecil Medicine, p. 1692
2. PATHOGENESIS
Step-by-step chain:
PIGA mutation
↓
No GPI anchor synthesis
↓
Loss of all GPI-linked complement regulatory proteins from cell surface:
• CD55 (DAF — Decay Accelerating Factor) → normally inactivates C3/C5 convertases
• CD59 (MIRL — Membrane Inhibitor of Reactive Lysis / Protectin) → normally blocks C9 polymerization
• C8-binding protein (homologous restriction factor)
↓
Complement activates unopposed (spontaneous, especially via alternative pathway)
↓
C5b-9 Membrane Attack Complex (MAC) assembles on RBC surface
↓
INTRAVASCULAR HEMOLYSIS
Why nocturnal / on waking?
During sleep → mild respiratory acidosis (CO₂ retention) → slight fall in blood pH → this activates complement → hemolysis peaks overnight → patient wakes and passes dark/cola-colored urine (hemoglobinuria). This is paroxysmal in only ~25% of cases; chronic low-grade hemolysis is the norm.
Why thrombosis?
- CD59 is also absent on platelets → platelet activation → externalization of phosphatidylserine → prothrombinase complex formation → hypercoagulability
- Free hemoglobin released into plasma scavenges nitric oxide (NO) → endothelial damage, platelet aggregation, smooth muscle contraction
RBC type classification by complement sensitivity:
| Type | GPI level | Complement sensitivity |
|---|
| Type I | Normal | Normal |
| Type II | Partial deficiency | 3–5× normal |
| Type III | Complete deficiency | 15–25× normal |
Coexistence of Type II + Type III in the same patient = two separate mutant clones.
— Robbins, Cotran & Kumar p. 606; Goldman-Cecil p. 1692; Henry's Clinical Diagnosis p. 693
3. CLINICAL FEATURES
Classic triad (remember: "HAT" — Hemolysis, Aplastic anemia risk, Thrombosis):
| Feature | Detail |
|---|
| Hemoglobinuria | Dark/cola urine in the morning (but present in minority; hemosiderinuria is almost always present) |
| Chronic hemolytic anemia | Variable severity, often mild-moderate |
| Thrombosis | ~40% of patients; leading cause of death; venous in 85% — hepatic veins (Budd-Chiari), portal veins, cerebral veins, abdominal veins |
| Abdominal pain | ~1/3 of patients; due to NO scavenging by free Hb |
| Dysphagia | Esophageal spasm; NO scavenging |
| Erectile dysfunction | Smooth muscle dysfunction; NO scavenging |
| Pancytopenia | Neutropenia in 3/5, thrombocytopenia in 2/3 at some point |
| Aplastic anemia evolution | ~1/3 of cases evolve into AA |
| AML transformation | Rare (~3%) |
| Splenomegaly | Uncommon |
| Hepatomegaly + ascites | Suggests intra-abdominal venous thrombosis |
Triggers for acute hemolytic episodes: infection, surgery, blood transfusion, contrast dye injection, severe exercise.
— Goldman-Cecil p. 1692; Henry's Clinical Diagnosis p. 693
4. LAB INVESTIGATIONS
Blood counts:
- Normocytic normochromic anemia (baseline)
- Can become hypochromic microcytic due to iron loss in urine (iron deficiency from chronic hemosiderinuria)
- Reticulocytosis — often less than expected for the degree of anemia
- Neutropenia (in ~60%)
- Thrombocytopenia (in ~66%)
- Pancytopenia is common
Hemolysis markers:
- ↑ LDH (marked elevation — intravascular hemolysis)
- ↓ Haptoglobin (consumed by free Hb)
- ↑ Indirect bilirubin
- Hemoglobinuria (urine dipstick positive for blood but no RBCs on microscopy)
- Hemosiderinuria — almost constantly present (iron in urine sediment; Prussian blue stain on urinary epithelial cells) → eventually causes iron deficiency
Coombs (DAT):
- Negative — critical distinguishing feature. PNH is a Coombs-negative intravascular hemolytic anemia. A DAT-negative hemolytic anemia + iron deficiency = think PNH
Bone marrow:
- May be hypercellular with erythroid hyperplasia
- Or hypocellular (when associated with AA)
Historical tests (now obsolete):
- Ham's test (Acidified serum lysis test) — RBCs lyse in acidified serum; replaced by flow cytometry
- Sucrose hemolysis test (Sugar water test) — RBCs lyse in low-ionic-strength sucrose solution; also replaced
Gold standard — Flow Cytometry:
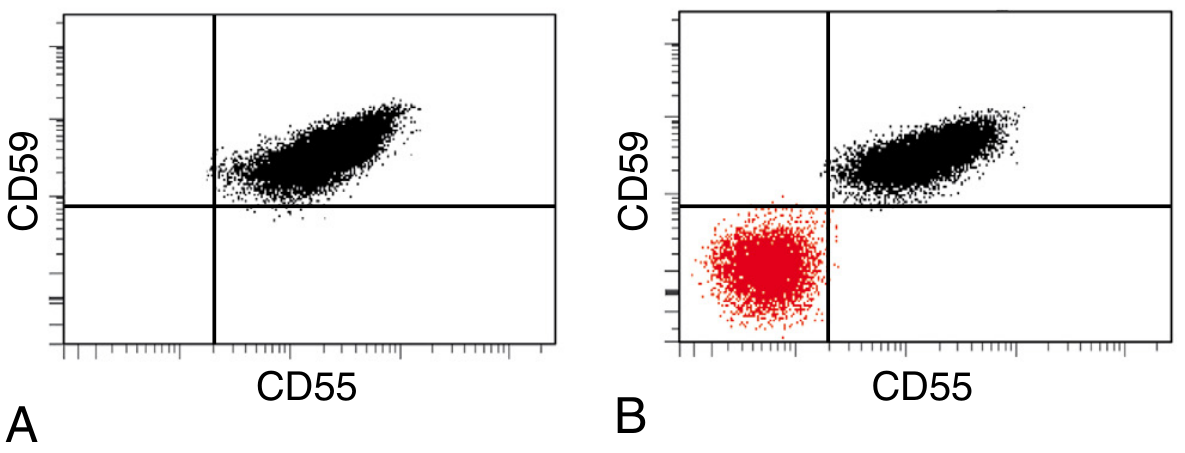
Fig: (A) Normal blood — all red cells express CD55 and CD59. (B) PNH blood — the red cluster (lower left) shows a population of RBCs completely lacking CD55 and CD59 — the PNH clone. — Robbins, Cotran & Kumar Pathologic Basis of Disease
What is tested:
- CD59 on RBCs (most sensitive for RBCs)
- CD55 and CD59 on RBCs
- CD24, CD57 on neutrophils; CD14 on monocytes
- FLAER (Fluorescent Aerolysin Variant) — bacterial toxin that binds GPI anchor directly; best and most reliable reagent for WBCs (neutrophils/monocytes); more sensitive than CD marker staining on leukocytes
Clone size correlates with degree of intravascular hemolysis.
— Goldman-Cecil p. 1692; Henry's Clinical Diagnosis p. 693; Robbins, Cotran & Kumar p. 606
5. PERIPHERAL SMEAR FINDINGS
PNH does not have pathognomonic smear findings (unlike sickle cell or spherocytosis), but you will see:
- Normocytic normochromic RBCs (baseline)
- Microcytic hypochromic RBCs if iron deficiency has supervened
- Polychromasia (reticulocytes)
- No spherocytes (unlike autoimmune hemolytic anemia)
- No sickling, no target cells
- The smear is often unremarkable — diagnosis is not made from the smear; it's made by flow cytometry
6. TREATMENT (for completeness)
Complement inhibitors (the main therapy):
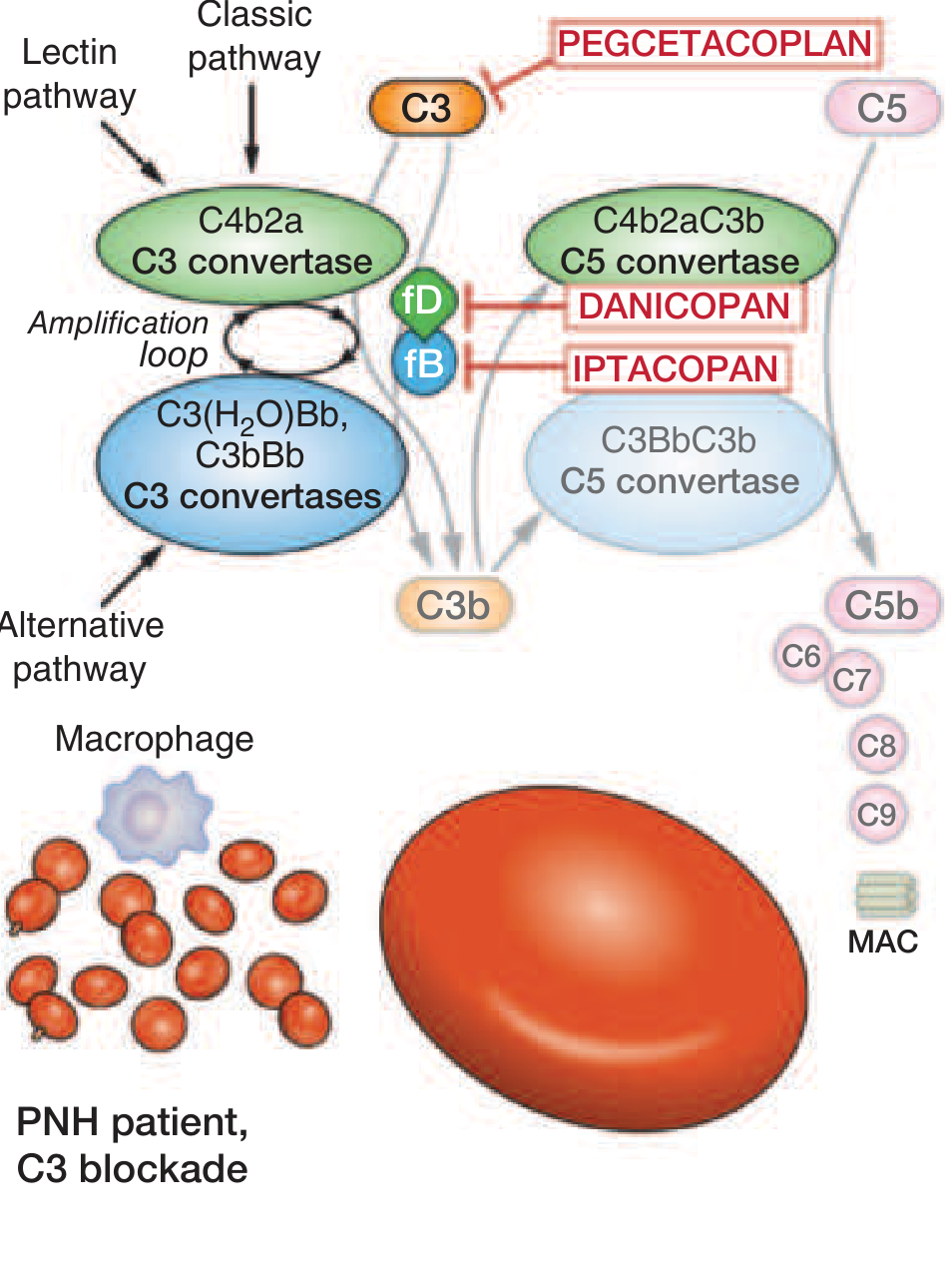
The complement cascade in PNH and sites of drug action. Eculizumab and ravulizumab block C5. Pegcetacoplan blocks C3. Danicopan and iptacoplan block factor D/B upstream. — Harrison's Principles of Internal Medicine 22E
| Drug | Target | Route | Notes |
|---|
| Eculizumab | C5 | IV q2 weeks | First approved; reduces intravascular hemolysis and thrombosis |
| Ravulizumab | C5 | IV q8 weeks | Longer half-life; equivalent efficacy |
| Pegcetacoplan | C3 | SC twice weekly | Blocks both intravascular AND extravascular hemolysis; corrects anemia more completely |
| Danicopan | Factor D | Oral | Used as add-on |
| Iptacoplan | Factor B/D | Oral | |
⚠ Before eculizumab: vaccinate against Meningococcus A, B, C (C5 blockade impairs terminal complement → high meningococcal risk). Long-term oral penicillin prophylaxis also recommended.
Note on eculizumab "paradox": C5 blockade prevents MAC → stops intravascular hemolysis. But PNH RBCs now accumulate C3b on surface → become opsonized → extravascular hemolysis by macrophages → DAT becomes positive. This is iatrogenic EV hemolysis.
Other management:
- Iron + folate supplementation (for chronic losses)
- Anticoagulation if thrombosis occurs
- Allogeneic BMT — only curative option; reserved for severe/refractory disease
- Transfusions as supportive care
- Eculizumab is indicated for transfusion-dependent hemolytic anemia or disease-related venous thrombosis
— Goldman-Cecil p. 1692; Harrison's 22E, p. 849
Quick Mental Recall Framework
PIGA mutation (X-linked, somatic, in HSC)
→ No GPI anchor
→ No CD55 / CD59 / C8-BP on RBCs, WBCs, Platelets
→ Complement runs unchecked → MAC → INTRAVASCULAR HEMOLYSIS
→ Nocturnal pH drop → triggers paroxysms
Clinical = Hemolysis + Thrombosis (unusual veins) + Pancytopenia ± AA
Urine = Hemoglobinuria + Hemosiderinuria → Iron deficiency
DAT = NEGATIVE (key!)
Diagnose = Flow cytometry (FLAER + CD59/CD55)
Treat = Eculizumab / Ravulizumab (C5 inhibitors) + vaccinate meningococcus
Cure = Allogeneic BMT
On your memory lapse: what happened is completely normal — this is the classic forgetting curve (Ebbinghaus). At 6 days with one revision at day 3, retention dropping to ~10% is expected without active retrieval practice. The fix is spaced repetition with active recall at shorter initial intervals (day 1 → day 3 → day 7 → day 14 → day 30), and each recall session should be self-testing, not re-reading. The framework above is built specifically so you can mentally walk through a cause → pathogenesis → clinical → lab → treatment chain rather than memorizing isolated facts.