Aspirin mechanism of action
Reading File
Finding Sources
Searching PubMed
aspirin mechanism action cyclooxygenase
Reading File
Reading File
Loading Image
Loading Image
Reading File
Here is a comprehensive breakdown of aspirin's mechanism of action, drawn from Lippincott's Pharmacology and Harrison's Principles of Internal Medicine.
Aspirin - Mechanism of Action
Core Biochemical Action
Aspirin (acetylsalicylic acid) is a weak organic acid that irreversibly acetylates the cyclooxygenase (COX) enzyme. It transfers its acetyl group to a serine residue (Ser-530 on COX-1, Ser-516 on COX-2) within the active site, permanently inactivating the enzyme. This makes aspirin unique among NSAIDs - all other NSAIDs are reversible inhibitors.
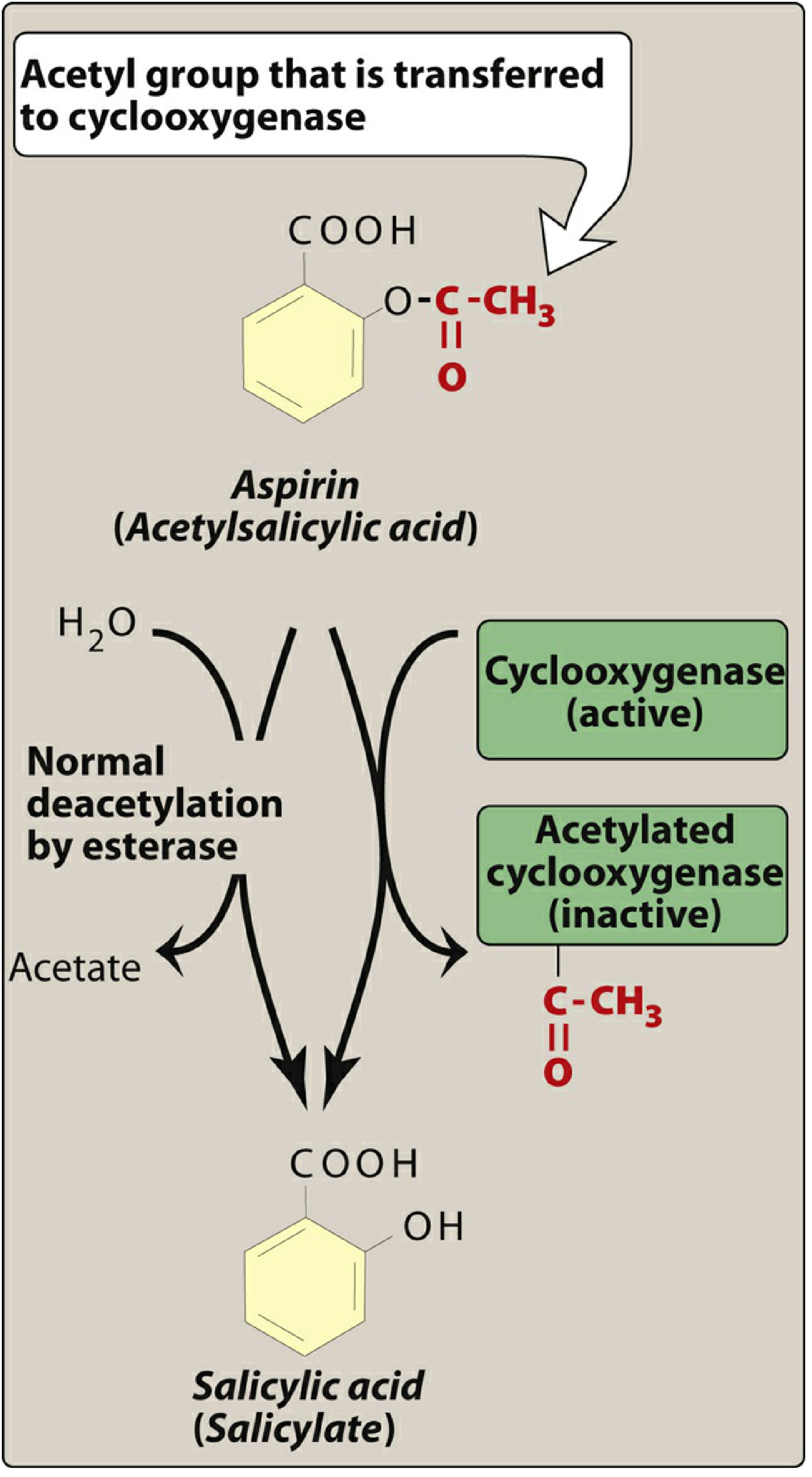
After acetylation, aspirin itself is hydrolysed to salicylic acid (the inactive metabolite) by plasma esterases.
The COX Pathway
COX enzymes (also called prostaglandin H2 synthase) catalyse the first step in prostanoid biosynthesis - converting arachidonic acid to prostaglandin H2 (PGH2), which is then further converted to various prostanoids:
| Prostanoid | Where made | Effect |
|---|---|---|
| Prostaglandins (PGE2, PGI2) | Widely distributed | Inflammation, pain sensitization, fever, vasodilation |
| Thromboxane A2 (TXA2) | Platelets (via COX-1) | Platelet aggregation, vasoconstriction |
| Prostacyclin (PGI2) | Endothelium (via COX-2) | Platelet inhibition, vasodilation |
Two COX Isoforms and Dose-Dependent Effects
| Feature | COX-1 | COX-2 |
|---|---|---|
| Expression | Constitutive (always present) | Inducible (upregulated in inflammation) |
| Location | Platelets, gastric mucosa, kidneys | Inflammatory cells, endothelium |
| Product in platelets | Thromboxane A2 | - |
| Product in endothelium | - | Prostacyclin (PGI2) |
Low-dose aspirin (75-325 mg/day):
- Selectively inhibits platelet COX-1 → reduces TXA2 synthesis → antiplatelet effect
- Platelets lack nuclei and cannot synthesize new COX, so inhibition lasts the platelet's entire lifespan (~7-10 days)
High-dose aspirin (~1 g/day):
- Also inhibits COX-2 in endothelial cells → reduces prostacyclin → this partially offsets the antithrombotic benefit
- Produces full anti-inflammatory, analgesic, and antipyretic effects
Three Major Therapeutic Actions (via COX inhibition)
1. Anti-inflammatory - Blocking COX diminishes prostaglandin formation, reducing prostaglandin-mediated inflammatory responses (vasodilation, vascular permeability, recruitment of leukocytes). Used in arthritis, but does not halt disease progression.
2. Analgesic - PGE2 sensitizes peripheral nociceptors to bradykinin, histamine, and other mediators. By reducing PGE2, aspirin raises the pain threshold. COX-2 inhibition is thought to be the primary driver of analgesia.
3. Antipyretic - Pyrogens (e.g., cytokines from activated WBCs) stimulate PGE2 synthesis, which raises the hypothalamic thermoregulatory set-point. Aspirin blocks PGE2 production, resetting the set-point to normal - causing peripheral vasodilation and sweating to dissipate heat. It has no effect on normal body temperature.
Antiplatelet Mechanism (in detail)
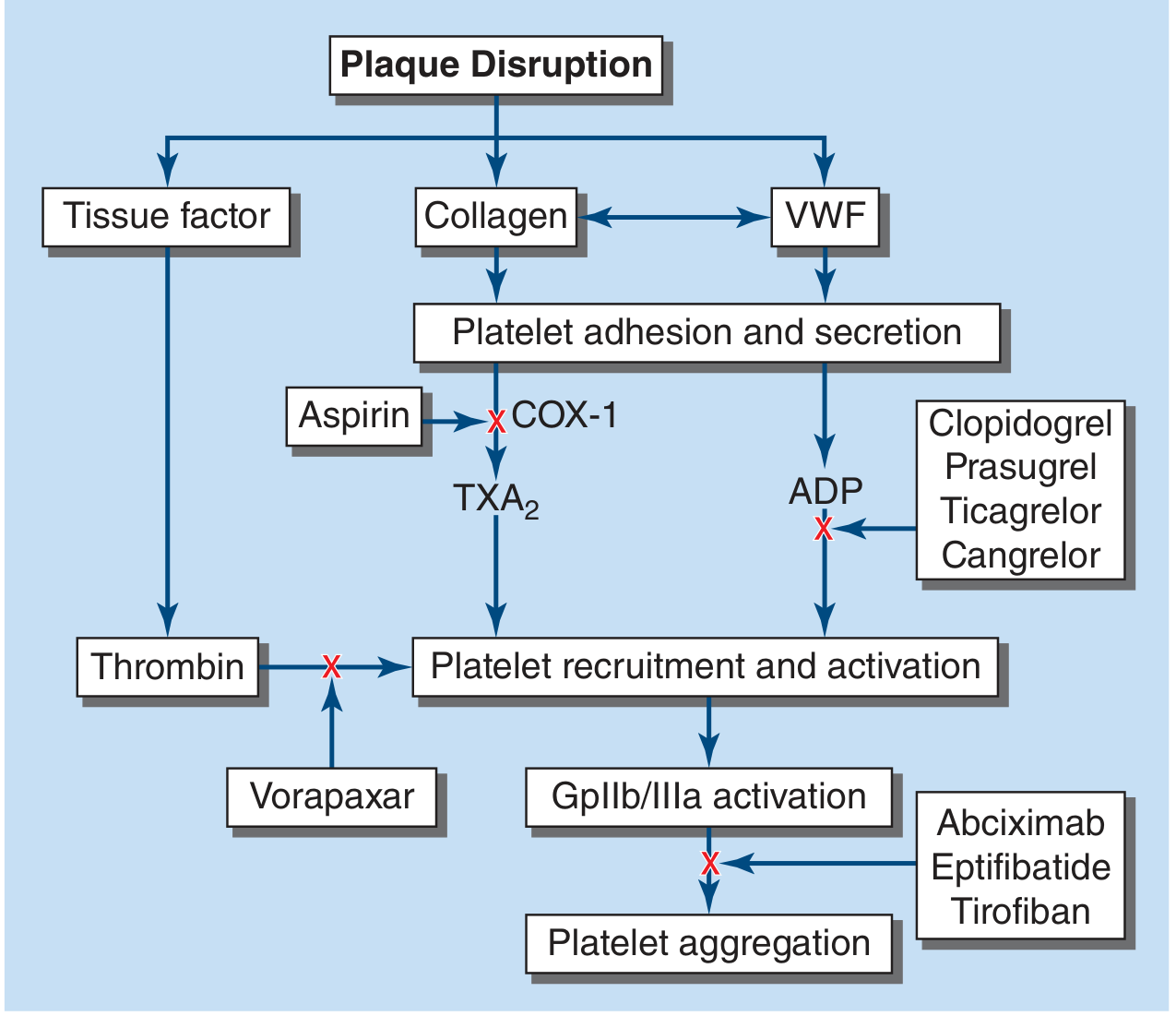
After plaque disruption, collagen and VWF trigger platelet adhesion. Aspirin blocks COX-1 in platelets, preventing TXA2 synthesis. Since TXA2 is a potent platelet activator and recruits further platelets to the thrombus, its suppression significantly reduces clot formation. This effect persists for the lifetime of the platelet because platelets cannot make new COX protein.
Key Pharmacological Distinctions
- Irreversibility - Unlike ibuprofen or naproxen (competitive/reversible inhibitors), aspirin's effect on platelets is permanent until new platelets are generated (recovery takes ~7-10 days after stopping aspirin)
- Dose-response relationship: Analgesic dose = ~650 mg QID; anti-inflammatory dose = 12-20 standard tablets/day; antiplatelet dose = 75-100 mg/day
- Aspirin-exacerbated respiratory disease (AERD): COX-1 inhibition in susceptible patients shifts arachidonic acid toward the lipoxygenase pathway, increasing leukotriene production - causing bronchospasm and nasal polyps
Sources: Lippincott Illustrated Reviews: Pharmacology, Ch. 40 | Harrison's Principles of Internal Medicine 22E, Ch. 123
This is a shared conversation. Sign in to Orris to start your own chat.