Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND mechanism
Finding Sources
Finding Sources
Searching Images
acute respiratory distress syndrome pathogenesis mechanism diffuse alveolar damage
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed for a comprehensive answer. Here is the complete explanation of ARDS mechanisms:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by severe diffuse lung inflammation, increased alveolar-capillary permeability, and hypoxemic respiratory failure. Its pathogenesis unfolds across several overlapping mechanisms.
1. Triggers and Initial Insult
ARDS arises from both direct (pulmonary) and indirect (extrapulmonary) insults:
| Direct Lung Injury | Indirect / Systemic Injury |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma / polytrauma |
| Pulmonary contusion | Pancreatitis |
| Near-drowning | Blood product transfusion (TRALI) |
| Inhalation injury | Burns, fat embolism |
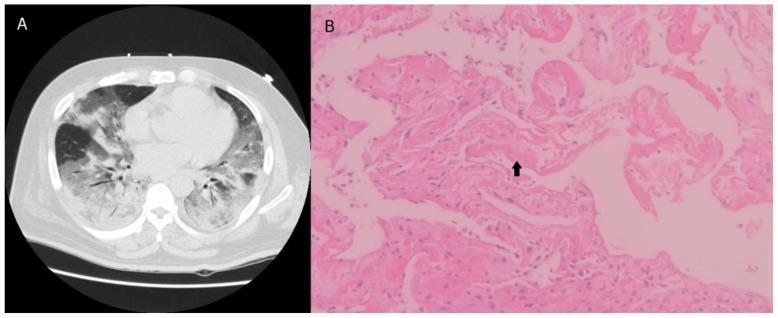
Regardless of the trigger, the final common pathway is diffuse alveolar damage (DAD).
2. Diffuse Alveolar Damage (DAD) — The Pathologic Hallmark
DAD evolves through three phases:
Exudative Phase (Days 1–7)
- Alveolar-capillary membrane disruption: Both the microvascular endothelium and alveolar epithelium (especially type I pneumocytes) are injured and killed (via necrosis, apoptosis, and mechanical stretch). Loss of this barrier allows protein-rich fluid to flood the alveolar space.
- Hyaline membrane formation: Leaked plasma proteins (fibrin, immunoglobulins) and cellular debris line the alveolar walls, forming the characteristic eosinophilic hyaline membranes seen on histology.
- Pulmonary edema: Increased permeability — not elevated hydrostatic pressure — is the defining feature distinguishing ARDS from cardiogenic pulmonary edema.
Proliferative Phase (Days 7–21)
- Type II pneumocytes proliferate in an attempt to resurface the denuded alveolar epithelium and restore barrier function.
- Fibroblast migration and early collagen deposition begin (elevated N-terminal procollagen III peptide can be detected in BAL fluid within 24 hours of onset).
Fibrotic Phase (>3 weeks)
- Some patients develop progressive fibrosis with architectural distortion, leading to chronic respiratory failure and an increased risk of barotrauma.
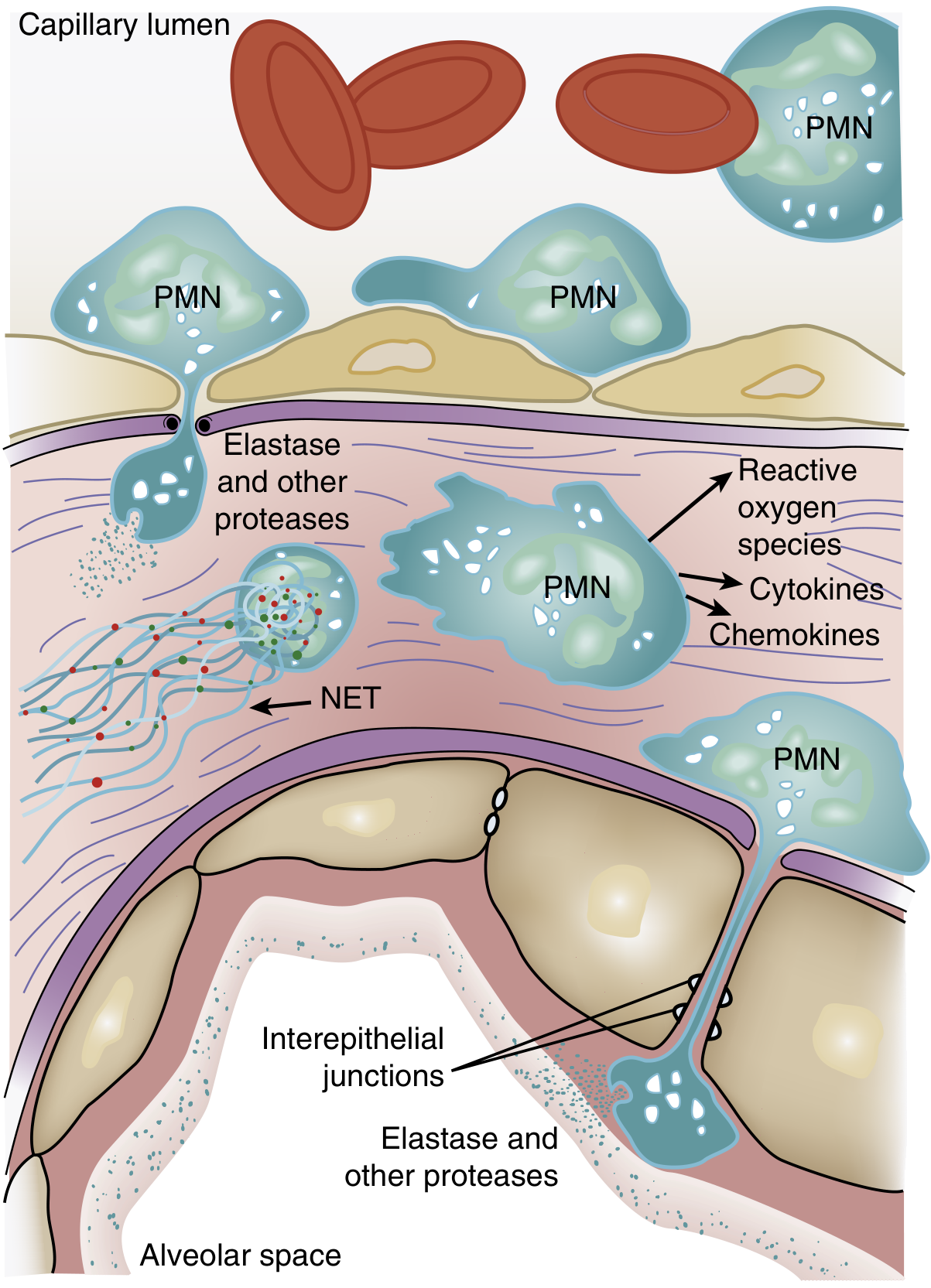
3. Neutrophil-Mediated Injury — Central Mechanism
Neutrophil accumulation in the pulmonary microvasculature is one of the earliest and most critical events in ARDS.
Sequestration mechanism:
- Pulmonary capillaries (~5 µm diameter) are narrower than neutrophils (~7–8 µm), forcing neutrophils to deform to transit the capillary bed. Activated, less deformable neutrophils become mechanically trapped.
- Simultaneously, upregulation of adhesion molecules (ICAM-1, E-selectin, P-selectin) on endothelial cells promotes neutrophil rolling and firm adhesion via integrin-ICAM interactions.
Cytokine cascade:
- Systemic inflammatory signals (TNF-α, IL-1β, IL-6, IL-8) from the primary insult activate circulating neutrophils and recruit them to the lung.
- IL-8 (a potent neutrophil chemoattractant) is markedly elevated in BAL fluid of ARDS patients.
Neutrophil effector mechanisms after transmigration:
- Reactive oxygen species (ROS): Superoxide, hydrogen peroxide, and hydroxyl radicals directly oxidize and damage endothelial and epithelial cell membranes.
- Proteolytic enzymes: Leukocyte elastase degrades the extracellular matrix (collagen, fibronectin, elastin) and also degrades surfactant protein A, amplifying alveolar dysfunction.
- Cytokines and chemokines: Activated neutrophils release TNF-α, IL-1β, and additional IL-8, amplifying the local inflammatory cascade.
- Neutrophil extracellular traps (NETs): Chromatin and granule proteins extruded from neutrophils contribute to microvascular thrombosis and further tissue injury.
4. Surfactant Dysfunction
- Injury to type II pneumocytes reduces synthesis of surfactant phospholipids (dipalmitoylphosphatidylcholine, phosphatidylglycerol).
- Leaked plasma proteins in the alveolar space inhibit surfactant function.
- Neutrophil elastase degrades surfactant protein A, further impairing the innate defense and surface tension reduction roles of surfactant.
- The ratio of large (biologically active) to small (inactive) surfactant aggregates is diminished.
- Consequence: Increased alveolar surface tension → diffuse microatelectasis → worsening ventilation-perfusion (V/Q) mismatch and intrapulmonary shunt.
5. Coagulation and Microvascular Thrombosis
- The alveolar space in ARDS becomes pro-coagulant: elevated tissue factor expression on injured epithelial cells, activated macrophages, and monocytes drives intra-alveolar fibrin deposition.
- Simultaneously, fibrinolysis is suppressed (elevated plasminogen activator inhibitor-1, PAI-1), leading to persistent fibrin clots that incorporate into hyaline membranes.
- Microvascular thrombosis compromises perfusion of lung units and can extend the zone of injury.
6. Impaired Alveolar Fluid Clearance
- Normal alveolar epithelium clears fluid by active sodium transport (via apical ENaC channels on type I/II pneumocytes and basolateral Na⁺/K⁺-ATPase), which drives osmotic water removal.
- In ARDS, epithelial injury impairs this sodium-driven fluid clearance — fluid accumulates faster than it can be removed.
- Clinical studies show that preserved or elevated alveolar fluid clearance is associated with improved survival, and impaired clearance predicts worse outcomes.
7. Angiopoietins and Endothelial Stability
- Angiopoietin-1 (Ang1) activates the Tie2 receptor on endothelium, stabilizing junctional integrity and reducing permeability.
- Angiopoietin-2 (Ang2) is stored in Weibel-Palade bodies and rapidly released upon endothelial activation; it antagonizes Tie2, promoting vascular leakage and inflammation.
- In ARDS, Ang2 levels are markedly elevated in plasma, correlating with severity and mortality. This Ang1/Ang2 imbalance is now a therapeutic target under investigation.
8. Ventilator-Induced Lung Injury (VILI)
Once a patient is mechanically ventilated, the lung injury can be perpetuated or worsened:
| Mechanism | Description |
|---|---|
| Volutrauma | Large tidal volumes overdistend non-dependent, aerated alveoli (the ARDS lung is heterogeneous — only ~30% is aerated). The same tidal volume inflates a "baby lung." |
| Barotrauma | Excessive plateau pressure disrupts alveolar walls → pneumothorax, pneumomediastinum |
| Atelectrauma | Repetitive collapse and reopening of alveoli generates high shear stress at the collapsed-aerated interface |
| Biotrauma | Mechanical stretch of cells triggers local cytokine release (IL-6, IL-8, TNF-α), extending inflammation systemically and contributing to multi-organ failure |
9. Net Physiologic Consequences
All of the above converge on three physiologic derangements:
- Intrapulmonary shunt (V/Q = 0): Fluid-filled, consolidated alveoli are perfused but not ventilated → severe hypoxemia refractory to supplemental O₂.
- Reduced lung compliance: Edema, atelectasis, and hyaline membranes stiffen the lung → high work of breathing, high driving pressures.
- Increased dead space (V/Q → ∞): Microvascular thrombosis and vasoconstriction reduce perfusion to ventilated units → hypercapnia in severe disease.
Summary Diagram (Conceptual)
Primary Insult (sepsis, pneumonia, aspiration, trauma)
↓
Systemic inflammatory activation (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration in pulmonary microvasculature
↓
Neutrophil transmigration → ROS + elastase + NETs + cytokines
↓
Endothelial + epithelial injury (Ang2 ↑, Tie2 destabilized)
↓
Alveolar-capillary barrier breakdown
↓
Protein-rich edema floods alveoli + hyaline membrane formation
↓
Surfactant dysfunction → microatelectasis
↓
V/Q mismatch + intrapulmonary shunt + ↓ compliance
↓
Hypoxemic respiratory failure (ARDS)
↓
VILI (if mechanically ventilated with injurious strategy)
↓
Biotrauma → multi-organ failure
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (ARDS: Etiology, Pathogenesis, Treatment)
- Goldman-Cecil Medicine, Chapter 91 (Mechanical Ventilation in ARDS)
- Sabiston Textbook of Surgery, Chapter on Critical Care
This is a shared conversation. Sign in to Orris to start your own chat.