Gout *** (clinc) Leschnyhan***(clinc) Purine denovo*** Purine salvage** Vit D Imp antioxidant vitamins B12 B9
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive content from the textbooks. Let me compose the complete answer.
Gout, Lesch-Nyhan, Purine Metabolism, Vitamin D, B12, B9 & Antioxidant Vitamins
1. GOUT ⭐⭐⭐ (Clinical)
Definition & Pathogenesis
Gout is marked by transient attacks of acute arthritis initiated by monosodium urate (MSU) crystals deposited within and around joints. It is characterized by hyperuricemia (plasma urate >6.8 mg/dL), which is necessary but not sufficient.
Uric acid accumulates by:
- Overproduction — enzymatic defects in purine synthesis (e.g., partial HGPRT deficiency), tumor lysis syndrome
- Underexcretion (~90% of primary gout) — reduced renal excretion; urate is filtered, almost completely reabsorbed by the proximal tubule, then a fraction is secreted distally
Types
| Primary Gout | Secondary Gout | |
|---|---|---|
| Cause | Reduced excretion (idiopathic) or enzymatic defects | Drugs (diuretics, cyclosporine), renal disease, myeloproliferative disorders, HGPRT deficiency |
| Proportion | ~90% | ~10% |
Mechanism of Acute Arthritis
- MSU crystals precipitate in joints
- Resident macrophages phagocytose crystals → activate the NLRP3 inflammasome → caspase-1 activation → IL-1β release
- Massive neutrophil recruitment → cytokines, free radicals, proteases, lysosomal enzyme release
- Results in acute arthritis → spontaneously remits in days to weeks
- Repeated attacks → tophi (aggregates of urate + inflammatory tissue) in synovial membrane and periarticular tissue → cartilage damage
Clinical Features
- Podagra — classic first attack: hot, red, swollen 1st metatarsophalangeal joint (MTP joint)
- Attacks often nocturnal, precipitated by: high-purine meals, alcohol, dehydration, surgery, trauma
- Tophi — in pinna of ear, Achilles tendon, periarticular tissue (after 20–30 years of hyperuricemia)
- Uric acid nephrolithiasis — radiolucent stones
- Gouty nephropathy — urate deposition in renal interstitium
Stages of Gout
- Asymptomatic hyperuricemia
- Acute gouty arthritis (intermittent attacks)
- Intercritical gout (asymptomatic between attacks)
- Chronic tophaceous gout
Risk Factors
- Male sex, postmenopausal women
- Obesity, hypertension, metabolic syndrome
- Alcohol (especially beer — high in guanosine)
- Medications: diuretics (thiazides, furosemide), cyclosporine, low-dose aspirin
- Chronic renal disease
Investigations
- Serum uric acid >6.8 mg/dL
- Joint aspiration (gold standard): needle-shaped, negatively birefringent MSU crystals (yellow when parallel to compensator)
- X-ray: "punched-out" erosions with overhanging edges ("rat-bite erosions"), soft-tissue tophi
- 24h urinary uric acid to distinguish overproducers vs. underexcreters
Management
Acute attack:
- Colchicine (first-line; inhibits microtubule polymerization → impairs neutrophil chemotaxis and inflammasome activation)
- NSAIDs (indomethacin preferred) — avoid in renal impairment
- Corticosteroids — if NSAIDs/colchicine contraindicated
Chronic/preventive (urate-lowering therapy — ULT):
- Target serum urate <6 mg/dL
- Allopurinol — xanthine oxidase inhibitor; start low (100 mg/day), titrate; avoid in azathioprine patients (or reduce AZA dose by 75%) because xanthine oxidase metabolizes azathioprine
- Febuxostat — non-purine selective xanthine oxidase inhibitor; 40–80 mg/day; no renal dose adjustment; ⚠️ higher CV mortality vs. allopurinol in high-risk patients
- Probenecid — uricosuric (blocks URAT1 renal transporter); avoid in nephrolithiasis
- Pegloticase — recombinant PEGylated uricase; converts uric acid → allantoin (soluble); for refractory gout; risk of antibody formation and infusion reactions
- Start ULT not during acute flare (can prolong attack); cover with colchicine prophylaxis when initiating
2. LESCH-NYHAN SYNDROME ⭐⭐⭐ (Clinical)
Genetics & Enzyme Defect
- X-linked recessive (affects males; females are carriers)
- Gene: HPRT1 on chromosome Xq26–q27
- Enzyme deficiency: Hypoxanthine-Guanine Phosphoribosyltransferase (HGPRT/HPRT)
- Complete (or near-complete) absence of HGPRT
Pathophysiology
- HGPRT catalyzes the salvage of hypoxanthine → IMP and guanine → GMP using PRPP
- Without HGPRT:
- Hypoxanthine is not recycled; instead degraded → xanthine → uric acid (overproduction hyperuricemia)
- PRPP accumulates (no longer consumed by salvage) → drives de novo purine synthesis further → more uric acid
- IMP and GMP levels fall → loss of feedback inhibition → amplified de novo synthesis
- CNS mechanism unclear but likely involves dopaminergic pathway dysfunction in basal ganglia
Clinical Features (onset ~3–6 months)
| Feature | Details |
|---|---|
| Self-mutilation | Compulsive biting of lips, fingers, tongue (pathognomonic) — begins year 2–3 |
| Neurological | Hypotonia → hypertonia, choreoathetosis, spasticity, tremor, dysarthria |
| Cognitive | Moderate intellectual disability |
| Hyperuricemia | Serum uric acid 7–10 mg/dL |
| Uric acid lithiasis | Orange-sand crystals in diapers (early sign) |
| Gouty tophi | Appear in ears after age 10 |
| Behavioral | Aggressiveness, compulsive actions |
Diagnosis
- Clinical features + elevated uric acid
- HGPRT enzyme assay (in RBCs)
- HPRT1 gene mutation analysis — accurate for carriers and affected males
Management
- Allopurinol — controls hyperuricemia and prevents uric acid nephropathy, but does NOT improve neurological/behavioral symptoms
- Behavioral management (physical restraints to prevent self-harm)
- Baclofen, benzodiazepines for spasticity
- Fluphenazine — reported to suppress self-mutilation when haloperidol failed
- No cure for neurological manifestations
Key Distinction: Partial vs. Complete HGPRT Deficiency
- Partial deficiency (Kelley-Seegmiller syndrome): hyperuricemia + gout, but no neurological features, no self-mutilation
- Complete deficiency: full Lesch-Nyhan syndrome
3. PURINE DE NOVO SYNTHESIS ⭐⭐⭐
Overview
- Synthesizes purine ring from scratch using small molecule precursors
- End product: IMP (inosine monophosphate)
- 11 steps, energetically expensive (multiple ATPs, glutamine, glycine, aspartate, folate derivatives consumed)
- Occurs primarily in the liver
Precursors of the Purine Ring
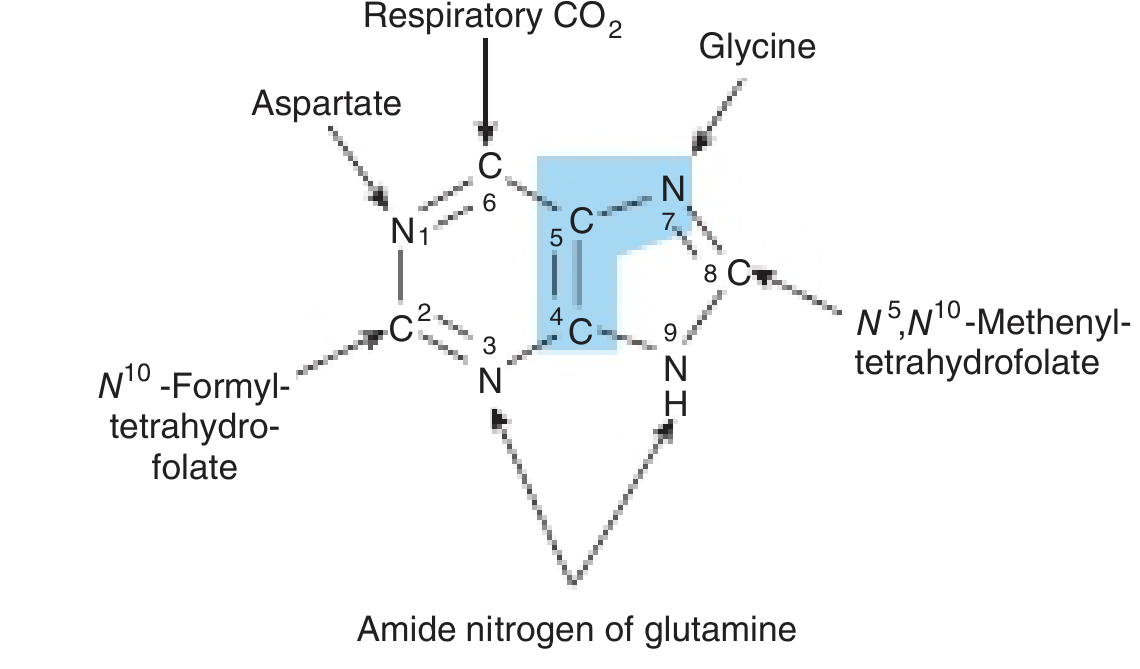
Key Steps
- PRPP synthesis (ribose-5-phosphate + 2 ATP → PRPP) by PRPP synthetase
- Glutamine donates amide N → 5-phosphoribosylamine (committed step, catalyzed by PRPP glutamyl amidotransferase)
- Sequential additions of glycine, formyl-THF (C8), glutamine (N3), CO₂ (C6), aspartate (N1), formyl-THF (C2) → closes the ring → IMP
- From IMP:
- IMP → AMP: adenylosuccinate synthetase (GTP-dependent) → adenylosuccinate lyase
- IMP → GMP: IMP dehydrogenase → GMP synthetase (ATP-dependent)
- Cross-regulation: AMP synthesis requires GTP; GMP synthesis requires ATP (mutual balance)
Regulation
- Rate-limiting factor: concentration of PRPP
- PRPP synthetase inhibited by AMP, ADP, GMP, GDP (feedback)
- PRPP glutamyl amidotransferase inhibited by AMP and GMP (feedback)
- Multifunctional enzymes in eukaryotes facilitate channeling of intermediates
Pharmacological Inhibition (anticancer/antifolate drugs)
- Methotrexate, aminopterin — inhibit dihydrofolate reductase → deplete THF → block steps 4 (C8 addition) and 10 (C2 addition)
- Azaserine, DON (6-diazo-5-oxo-L-norleucine) — glutamine analogs → block steps 2, 5, 8
- Mycophenolate mofetil (MMF) — inhibits IMP dehydrogenase → blocks de novo GMP synthesis → selectively affects T and B lymphocytes (which lack efficient salvage pathway)
4. PURINE SALVAGE PATHWAY ⭐⭐
Overview
- Recycles free purine bases (from diet or nucleic acid degradation) back into nucleotides
- Far less energy-intensive than de novo synthesis
- Particularly important in tissues with low de novo capacity: brain, RBCs, PMNs
Key Enzymes
| Enzyme | Reaction | Clinical Relevance |
|---|---|---|
| HGPRT (hypoxanthine-guanine phosphoribosyl transferase) | Hypoxanthine + PRPP → IMP; Guanine + PRPP → GMP | Deficient in Lesch-Nyhan |
| APRT (adenine phosphoribosyl transferase) | Adenine + PRPP → AMP | Deficiency → 2,8-dihydroxyadenine stones |
| Adenosine kinase | Adenosine + ATP → AMP + ADP | — |
| Deoxycytidine kinase | Phosphorylates dCyd, dGuo → dCMP, dGMP | — |
Mechanism
Purine base + PRPP → Purine-5'-monophosphate + PPi
(phosphoribosylation)
Regulation Feedback
- Salvage-generated AMP, GMP, IMP feedback inhibit PRPP synthetase and PRPP glutamyl amidotransferase, suppressing de novo synthesis
- This is why HGPRT deficiency → ↑ free PRPP → ↑ de novo synthesis → ↑ uric acid
Clinical Relevance
- Lesch-Nyhan syndrome — HGPRT deficiency (see above)
- ADA deficiency (adenosine deaminase) — accumulation of dATP inhibits ribonucleotide reductase → severe combined immunodeficiency (SCID)
- PNP deficiency (purine nucleoside phosphorylase) — dGTP accumulates → T-cell deficiency, normal B cells
- Azathioprine — prodrug metabolized to 6-mercaptopurine → incorporated via salvage → inhibits de novo purine synthesis → immunosuppression
5. VITAMIN D
Synthesis & Activation (Two-Step Hydroxylation)
7-Dehydrocholesterol (skin)
↓ UV light (290–320 nm)
Previtamin D3 → Vitamin D3 (cholecalciferol) [enters dermal capillaries]
↓ Liver — 25-hydroxylase
25-hydroxyvitamin D [25(OH)D3] — storage form, measured in serum
↓ Kidney — 1α-hydroxylase (regulated by PTH, hypophosphatemia)
1,25-dihydroxyvitamin D3 = Calcitriol (active form)
- Dietary sources: fortified milk, fatty fish (salmon, sardines, tuna, cod), fish oil
- Ergocalciferol (D2) from plant sources joins the same pathway at the liver step
Actions of Calcitriol (1,25(OH)₂D₃)
- Intestine: ↑ calcium and phosphate absorption (nuclear receptor → transcription of calbindin)
- Bone: stimulates osteoclast-mediated bone resorption → releases Ca²⁺ and PO₄
- Kidney: ↑ tubular reabsorption of Ca²⁺ and phosphate
- Immune: activates macrophages via Toll-like receptor-triggered VDR expression; protective against TB
Deficiency — Rickets/Osteomalacia
- Children: Rickets — failure of bone mineralization, bowing of long bones, rachitic rosary, craniotabes
- Adults: Osteomalacia — bone pain, proximal myopathy, insufficiency fractures
High-risk groups: exclusively breastfed dark-skinned infants, elderly housebound, malabsorption (celiac, Crohn, post-gastrectomy), anticonvulsant use (induces hepatic catabolism), chronic renal failure, dark-skinned individuals in low-sun areas
Genetic Disorders of Vitamin D Metabolism
| Type | Defect | Treatment |
|---|---|---|
| Vitamin D-Dependent Rickets Type I (VDDR-I) | Autosomal recessive; deficient renal 1α-hydroxylase | Calcitriol supplementation |
| Vitamin D-Dependent Rickets Type II (VDDR-II) | Autosomal recessive; end-organ resistance (VDR mutation) | High-dose calcitriol + calcium |
Emerging Associations
- ↓ vitamin D linked to: hypertension, ↑ fasting glucose/insulin, cardiovascular disease, hip fractures, colon cancer mortality, ↑ all-cause mortality
6. IMPORTANT ANTIOXIDANT VITAMINS
Vitamin C (Ascorbic Acid)
- Water-soluble antioxidant
- Directly scavenges ROS (superoxide, hydroxyl radicals, singlet oxygen)
- Regenerates vitamin E (reduces tocopheroxyl radical back to tocopherol)
- Co-factor for prolyl and lysyl hydroxylase (collagen synthesis) → deficiency = scurvy
- Scurvy: perifollicular hemorrhages, corkscrew hairs, gingival bleeding/swelling, impaired wound healing
Vitamin E (Tocopherols/Tocotrienols)
- Fat-soluble antioxidant — major lipid-phase antioxidant
- α-tocopherol = most biologically active form
- Protects polyunsaturated fatty acids (PUFAs) in cell membranes from lipid peroxidation
- Mechanism: donates H· to lipid peroxy radical → chain-breaking antioxidant
- Deficiency: hemolytic anemia, spinocerebellar ataxia (in children with fat malabsorption), peripheral neuropathy
- Found in vegetable oils, nuts, seeds, green leafy vegetables
7. VITAMIN B12 (COBALAMIN) ⭐⭐
Structure & Sources
- Cobalt-containing vitamin; animal products only (meat, liver, dairy, eggs)
- Plant-based diets → risk of deficiency without supplementation
Absorption
- Requires Intrinsic Factor (IF) — secreted by gastric parietal cells (Castle's intrinsic factor)
- IF-B12 complex → absorbed at terminal ileum
- Transported by transcobalamin II in plasma
Causes of Deficiency
- Pernicious anemia — autoimmune destruction of parietal cells → ↓ IF → B12 malabsorption
- Gastrectomy, ileal resection, Crohn's disease (terminal ileum)
- Strict veganism
- Drugs: metformin (↓ IF-B12 absorption), prolonged PPI use, nitrous oxide (oxidizes cobalamin)
Functions
- Methylmalonyl-CoA mutase — converts methylmalonyl-CoA → succinyl-CoA (requires adenosylcobalamin)
- Deficiency → ↑ methylmalonic acid (MMA) — marker of B12 deficiency
- Methionine synthase — converts homocysteine → methionine (requires methylcobalamin); regenerates THF from methyl-THF ("methyl-folate trap")
- Deficiency → ↑ homocysteine; impaired myelin synthesis
Clinical Features of Deficiency
- Hematologic: macrocytic megaloblastic anemia (impaired DNA synthesis in RBCs), hypersegmented neutrophils
- Neurological (subacute combined degeneration of spinal cord): demyelination of posterior columns (vibration/position sense loss) + lateral corticospinal tracts (UMN signs); peripheral neuropathy
- NB: Folate deficiency also causes megaloblastic anemia but NO neurological features
Diagnosis
- ↑ MCV; megaloblastic changes on blood smear
- ↓ Serum B12
- ↑ Serum methylmalonic acid (most sensitive) + ↑ homocysteine
Treatment
- IM cyanocobalamin or hydroxocobalamin (if absorption defective)
- Oral high-dose B12 (1000 µg/day) if absorption intact
8. VITAMIN B9 (FOLATE) ⭐⭐
Sources & Absorption
- Liver, leafy green vegetables, legumes, wheat bran, fortified cereals
- Absorbed in jejunum (proximal small bowel)
- Body stores last only 3–4 months (unlike B12 which lasts years)
Active Form
- Tetrahydrofolate (THF) — carrier of single-carbon units
- Used in: purine synthesis (C2, C8 of purine ring), thymidylate synthesis (dUMP → dTMP), amino acid metabolism (serine ↔ glycine interconversion, homocysteine → methionine)
Causes of Deficiency
- Poor diet (alcoholism, poverty)
- Malabsorption: celiac disease, chronic diarrhea, total gastrectomy
- Antifolate drugs:
- Methotrexate, trimethoprim, pyrimethamine (inhibit dihydrofolate reductase)
- Oral contraceptives
- Antiepileptics (phenobarbital, phenytoin — induce hepatic enzymes, deplete folate)
- Increased demand: pregnancy, hemolytic anemia, malignancy
Clinical Features
- Megaloblastic anemia + hypersegmented neutrophils (same as B12)
- Glossitis (smooth tongue), angular cheilitis, mucosal ulcers
- Brown hyperpigmentation in palmar creases and flexures
- No neurological symptoms (key distinction from B12 deficiency)
- Neural tube defects (spina bifida, anencephaly) — with periconceptional deficiency
Treatment
- Folic acid 1–5 mg/day orally
- ⚠️ Must rule out B12 deficiency first — folate supplementation corrects anemia but allows neurological damage from B12 deficiency to progress undetected ("masking")
Supplementation
- All women of childbearing age: 400 µg/day folic acid to prevent neural tube defects
- High-risk women (prior NTD, antiepileptic use): 4–5 mg/day
Quick Summary Table
| Topic | Key Enzyme/Mechanism | Clinical Consequence |
|---|---|---|
| Gout | ↑ Uric acid → MSU crystals → NLRP3 inflammasome → IL-1β | Acute arthritis, tophi, nephropathy |
| Lesch-Nyhan | HGPRT absent → ↑ PRPP → ↑ uric acid | Self-mutilation, choreoathetosis, hyperuricemia |
| De novo purine | PRPP → IMP (11 steps); needs THF, glycine, glutamine, aspartate | Blocked by methotrexate, MMF |
| Salvage purine | HGPRT/APRT recycle free purines + PRPP → nucleotides | HGPRT deficiency = Lesch-Nyhan; ADA deficiency = SCID |
| Vitamin D | 7-DHC → D3 → 25(OH)D (liver) → 1,25(OH)₂D (kidney) | Rickets (children), osteomalacia (adults) |
| Vitamin E | Lipid-phase antioxidant; breaks lipid peroxidation chains | Hemolytic anemia, spinocerebellar ataxia |
| Vitamin C | Water-phase antioxidant; regenerates Vit E; collagen | Scurvy |
| Vitamin B12 | Methylmalonyl-CoA mutase + methionine synthase | Megaloblastic anemia + subacute combined degeneration |
| Vitamin B9 | THF → single-carbon transfers for purine/dTMP synthesis | Megaloblastic anemia (no neuro), neural tube defects |
Sources: Robbins & Kumar Basic Pathology; Harper's Illustrated Biochemistry 32nd Ed; Adams & Victor's Principles of Neurology 12th Ed; Fitzpatrick's Dermatology; Comprehensive Clinical Nephrology 7th Ed
This is a shared conversation. Sign in to Orris to start your own chat.