myopathies diagnosis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the content needed to deliver a comprehensive, well-structured answer on myopathy diagnosis.
Diagnosis of Myopathies
Myopathies are primary disorders of skeletal muscle. Diagnosis integrates clinical presentation, laboratory investigations, electrodiagnostics, and muscle biopsy — each layer narrowing the differential.
1. Clinical Approach
Key clinical features suggesting a myopathy (vs. neuropathy):
- Proximal > distal weakness (difficulty rising from chair, climbing stairs, raising arms overhead)
- No sensory symptoms
- Preserved reflexes early
- Characteristic skin lesions (dermatomyositis)
History clues:
- Onset (acute, subacute, chronic/lifelong)
- Family history → hereditary myopathy
- Drug use (statins, glucocorticoids, colchicine, zidovudine, immune checkpoint inhibitors)
- Systemic illness, connective tissue disease
2. Classification Framework
The four major subtypes of inflammatory myopathy have distinct clinical and pathological features:
| Disorder | Age | Clinical Features | Biopsy |
|---|---|---|---|
| Dermatomyositis | Juvenile + adult | Proximal weakness + skin rash | Perimysial/perivascular inflammation; perifascicular atrophy (pathognomonic) |
| Polymyositis | Adult (rare in childhood) | Proximal weakness | Endomysial inflammation, non-specific |
| Immune-mediated necrotizing myopathy | Adult | Proximal weakness | Scattered necrotic/regenerating myofibers; macrophage invasion |
| Inclusion body myositis (IBM) | Adult >40 years | Finger flexor + quadriceps weakness; treatment-refractory | Endomysial inflammation + invasion of non-necrotic fibers; rimmed vacuoles |
— Goldman-Cecil Medicine, p. 2844
3. Laboratory Investigations
Serum Enzymes
- Creatine kinase (CK): Most sensitive marker; elevated in most myopathies. Can be normal in dermatomyositis (even below normal in highly active disease) and in some toxic/metabolic myopathies.
- Aldolase: May be elevated when CK is normal (especially dermatomyositis).
- LDH, AST, ALT may also be elevated.
Autoantibodies (Myositis-Specific)
- Anti-Jo-1 (anti-histidyl-tRNA synthetase): polymyositis/dermatomyositis; associated with interstitial lung disease and "mechanic's hands"
- Anti-Mi-2, Anti-MDA5: dermatomyositis
- Anti-HMGR (HMG-CoA reductase): immune-mediated necrotizing myopathy (statin-associated)
- Anti-SRP: immune-mediated necrotizing myopathy; may be paraneoplastic
- Anti-cN1A (NT5C1A): present in 50–70% of IBM patients; >90–95% specific for IBM among muscle diseases
Malignancy Screening
Required in dermatomyositis and anti-SRP myopathy: CBC, urinalysis, chemistries, LFTs, CXR, stool occult blood, mammogram (women), PSA (men) + age/gender-appropriate cancer screening.
4. Electrodiagnostics (EMG/NCS)
Nerve conduction studies (NCS): Normal in most primary myopathies (helps exclude neuropathy).
Needle EMG in myopathy shows:
- Small, short-duration, polyphasic motor unit action potentials (MUAPs)
- Early recruitment (more MUAPs for a given force)
- Abnormal spontaneous activity (fibrillations, positive sharp waves) in inflammatory/necrotizing myopathies
In critical illness myopathy: muscles become unresponsive to direct electrical stimulation — a distinguishing feature from prolonged neuromuscular blockade.
The diagnosis of inflammatory myopathy requires exclusion of electrolyte abnormalities (hypokalemia, hypophosphatemia). An elevated CK alone does not establish a myopathic cause, since some neuropathies also raise CK. — Rosen's Emergency Medicine
5. Muscle Biopsy — The Diagnostic Gold Standard
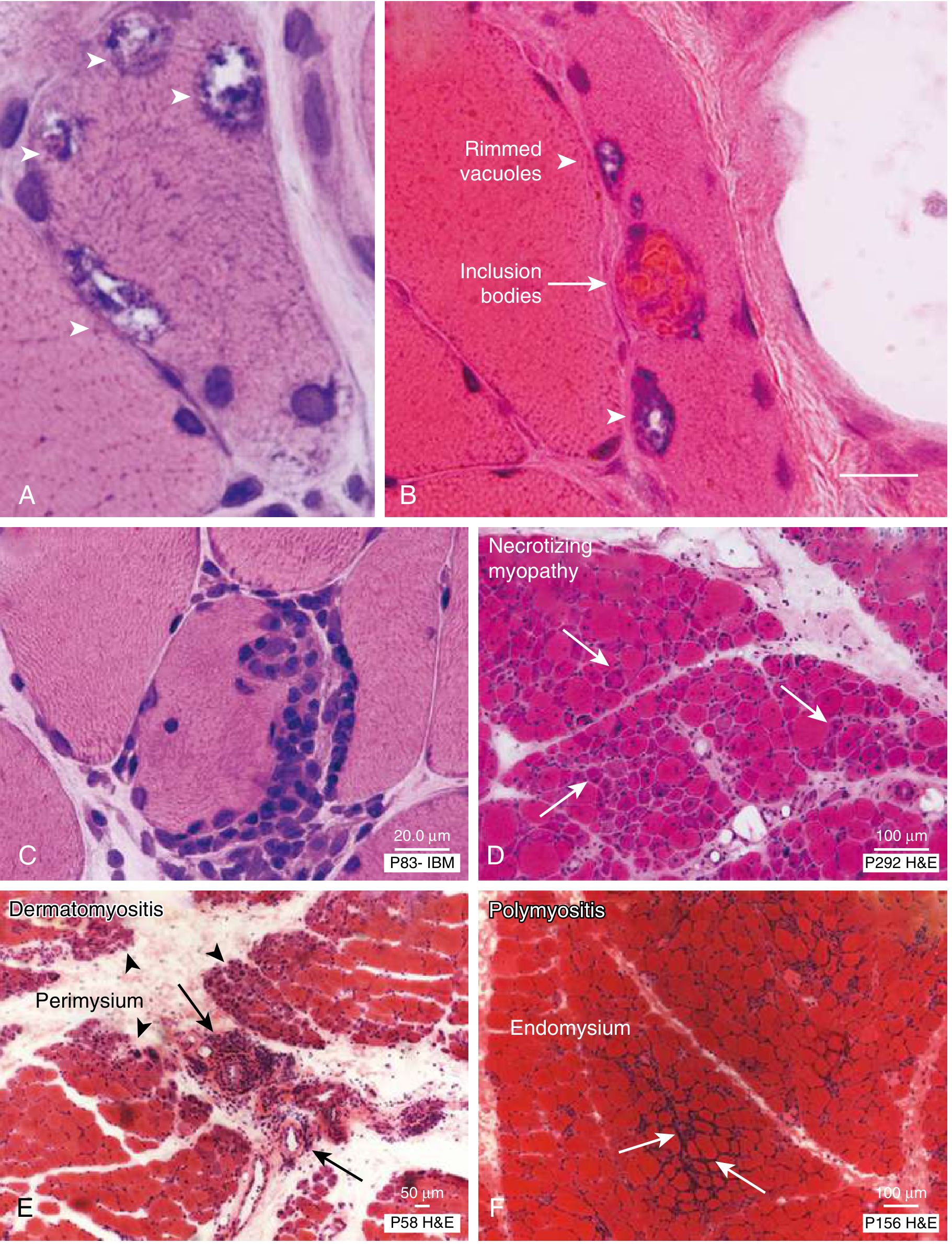
A–B: Rimmed vacuoles and inclusion bodies in IBM. C: Invasion of non-necrotic fiber (IBM). D: Scattered necrotic/regenerating fibers in necrotizing myopathy. E: Perimysial inflammation with perifascicular atrophy in dermatomyositis. F: Endomysial inflammation in polymyositis.
Key biopsy findings by disease:
| Disease | Pathognomonic/Characteristic Finding |
|---|---|
| Dermatomyositis | Perifascicular atrophy (pathognomonic); perimysial & perivascular inflammation |
| Polymyositis | Endomysial inflammation; CD8+ T-cell invasion of non-necrotic fibers |
| IBM | Rimmed vacuoles; endomysial inflammation; invasion of non-necrotic fibers |
| Necrotizing myopathy | Necrotic/regenerating fibers; macrophage infiltration; minimal lymphocytic inflammation |
| Toxic/metabolic myopathy | Variable; type II fiber atrophy; myosin filament loss (thick-filament myopathy) |
For dermatomyositis, skin biopsy showing cell-poor interface dermatitis supports the diagnosis and may avoid muscle biopsy.
6. Imaging
MRI of muscle: Increasingly used to:
- Identify active inflammatory changes (STIR sequences — edema/inflammation)
- Guide biopsy site selection
- Assess disease distribution (distal vs. proximal, symmetric vs. asymmetric)
7. Diagnostic Approach by Subtype
Dermatomyositis
Diagnosis supported by:
- Characteristic skin findings: heliotrope rash (violaceous periorbital erythema) ± edema, Gottron papules (violaceous papules over dorsal MCP/PIP joints)
- Proximal weakness
- Elevated CK (or normal/low)
- Positive myositis-specific antibodies (anti-Mi-2, anti-MDA5, anti-Jo-1)
- Biopsy: perifascicular atrophy (pathognomonic)
Polymyositis
Core diagnostic criteria:
- Subacute proximal weakness
- Elevated serum CK
- Biopsy: endomysial inflammation without features of IBM or other specific diagnosis ⚠️ Diagnosis is problematic — limb-girdle muscular dystrophy and IBM are frequently misdiagnosed as polymyositis.
Inclusion Body Myositis (IBM)
Key features:
- Age >50 years; slowly progressive (years, not months)
- Asymmetric finger flexor and quadriceps weakness
- ~50% of patients are initially misdiagnosed (often as polymyositis)
- Anti-cN1A antibodies (50–70% sensitivity; >90–95% specificity)
- Biopsy: rimmed vacuoles, endomysial inflammation, invasion of non-necrotic fibers
- Does not respond to immunosuppression (distinguishing from polymyositis)
Immune-Mediated Necrotizing Myopathy (IMNM)
- Acute/subacute proximal weakness + very high CK
- Anti-HMGR antibodies (statin-associated) or anti-SRP antibodies
- Biopsy: necrotic/regenerating fibers with macrophages but minimal lymphocytic inflammation
- Screen for malignancy (paraneoplastic, esp. with anti-SRP)
8. Toxic & Metabolic Myopathies
Common causes requiring exclusion:
| Category | Agents |
|---|---|
| Rhabdomyolysis/chronic myopathy | Statins (lovastatin, simvastatin, atorvastatin), fibrates, alcohol, cocaine, zidovudine, cyclosporine |
| Painful myopathy ± rhabdomyolysis | Colchicine, emetine, taxanes |
| Inflammatory myopathy (drug-induced) | Hydralazine, penicillamine, procainamide, immune checkpoint inhibitors (esp. anti-PD-1), statins (HMGR-antibody) |
| Myalgia ± myopathy | Amiodarone, aromatase inhibitors, bisphosphonates, calcium channel blockers, fluoroquinolones, beta-blockers |
CK, EMG, and biopsy may all be normal in some toxic myopathies; diagnosis depends on drug history and resolution with drug withdrawal. — Bradley & Daroff's Neurology, p. 1667
Summary: Diagnostic Checklist
| Step | Test |
|---|---|
| 1 | Serum CK, aldolase, LDH |
| 2 | Myositis-specific antibodies panel |
| 3 | Thyroid function (hypothyroid myopathy mimic) |
| 4 | EMG/NCS |
| 5 | MRI muscle (distribution, guide biopsy) |
| 6 | Muscle biopsy (H&E, modified Gomori trichrome, ATPase, immunostaining) |
| 7 | Skin biopsy (if dermatomyositis suspected) |
| 8 | Malignancy screening (dermatomyositis, anti-SRP IMNM) |
| 9 | Genetic testing (if hereditary myopathy suspected: dystrophinopathies, LGMD, metabolic myopathies) |
— Goldman-Cecil Medicine, pp. 2843–2845 | Bradley & Daroff's Neurology | Rosen's Emergency Medicine
This is a shared conversation. Sign in to Orris to start your own chat.