myopathies
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have enough comprehensive content for a high-level overview. Let me synthesize a well-organized summary.
Myopathies — Overview & Classification
Myopathies are disorders characterized by structural changes or functional impairment of muscle, distinct from other motor unit diseases (anterior horn cell disease, neuropathies, neuromuscular junction disorders) by clinical and laboratory features. — Harrison's Principles of Internal Medicine, 22e
Key Clinical Features
Typical presentation:
- Proximal, symmetric limb weakness — difficulty rising from a chair, climbing stairs, raising arms overhead (waddling gait, Gowers' sign)
- Preserved deep tendon reflexes (vs. neuropathy)
- No sensory loss (sensory loss points to peripheral neuropathy or CNS lesion)
- No fasciculations (which suggest motor neuron disease)
Weakness patterns that help narrow the differential:
| Pattern | Condition to Consider |
|---|---|
| Limb-girdle (proximal > distal, symmetric) | Muscular dystrophies, inflammatory myopathies |
| Facial + scapular winging | Facioscapulohumeral dystrophy (FSHD) |
| Facial + distal + grip myotonia | Myotonic dystrophy type 1 |
| Ptosis + ophthalmoparesis | Oculopharyngeal dystrophy, mitochondrial myopathy |
| Finger/wrist flexor + quadriceps (asymmetric) | Inclusion body myositis (IBM) |
| Episodic weakness + myoglobinuria | Metabolic myopathies (glycogenoses, lipid disorders) |
Classification
1. Muscular Dystrophies
Progressive hereditary disorders with muscle fiber degeneration and replacement by fat/connective tissue.
- Duchenne (DMD) / Becker (BMD) — X-linked; dystrophin gene mutations; childhood onset; pseudohypertrophy of calves; DMD is severe/progressive, BMD is milder
- Emery-Dreifuss (EDMD) — X-linked or AD/AR; early contractures (elbow, Achilles), cardiac involvement (arrhythmias) obligatory
- Limb-Girdle Muscular Dystrophies (LGMD) — Autosomal dominant or recessive; affect pelvic and shoulder girdle; genetically heterogeneous (sarcoglycanopathies, calpainopathy, dysferlinopathy, etc.)
- Facioscapulohumeral (FSHD) — AD; facial, scapular, and humeral weakness; often asymmetric
- Oculopharyngeal — AD; ptosis and dysphagia in mid-adulthood
2. Myotonic Myopathies
Defined by myotonia — prolonged muscle contraction with slow relaxation (grip myotonia, percussion myotonia).
- Myotonic Dystrophy Type 1 (DM1) — DMPK gene (CTG repeat); distal > proximal weakness; multisystem (cataracts, arrhythmia, endocrine, cognitive)
- Myotonic Dystrophy Type 2 (DM2) — CNBP gene; proximal > distal
- Myotonia Congenita — CLCN1 (chloride channel); myotonia without significant weakness; muscle hypertrophy
- Paramyotonia Congenita — SCN4A (sodium channel); paradoxical worsening with repeated activity; cold-triggered
3. Inflammatory Myopathies (Idiopathic)
Immune-mediated; elevated CK; inflammatory infiltrates on biopsy.
| Subtype | Key Features |
|---|---|
| Dermatomyositis (DM) | Proximal weakness + skin findings (heliotrope rash, Gottron's papules); perimysial/perivascular inflammation; associated with malignancy |
| Polymyositis (PM) | Proximal weakness without skin changes; endomysial T-cell infiltration; largely a diagnosis of exclusion |
| Inclusion Body Myositis (IBM) | Most common >50 yrs; distal + proximal, asymmetric; finger/wrist flexors + quads; poor response to immunotherapy |
| Immune-Mediated Necrotizing Myopathy (IMNM) | Necrosis with minimal inflammation; anti-SRP or anti-HMGCR antibodies; statin-associated |
| Antisynthetase Syndrome | Anti-Jo-1 and other anti-tRNA synthetase antibodies; ILD, inflammatory arthritis, mechanic's hands |
4. Metabolic Myopathies
Inherited defects in energy metabolism → episodic weakness/myoglobinuria or fixed weakness.
- Glycogen storage diseases — McArdle disease (myophosphorylase deficiency): exercise intolerance, cramps, "second wind" phenomenon; forearm exercise test shows no lactate rise
- Lipid disorders — CPT2 (carnitine palmitoyltransferase 2) deficiency: prolonged low-intensity exercise triggers rhabdomyolysis; fasting/cold are triggers
- Mitochondrial myopathies — Maternal inheritance (mtDNA mutations); chronic progressive external ophthalmoplegia (CPEO), MELAS, MERRF, Kearns-Sayre syndrome; ragged red fibers on Gomori trichrome stain
5. Congenital Myopathies
Present at birth or early childhood; structural abnormalities on biopsy; often non-progressive.
| Type | Biopsy Finding |
|---|---|
| Nemaline myopathy | Nemaline (rod) bodies |
| Central core disease | Central cores lacking oxidative activity |
| Centronuclear / Myotubular myopathy | Centrally placed nuclei |
| Multicore disease | Multiple small cores |
6. Channelopathies / Ion Channel Disorders
- Periodic paralyses — Hypokalemic (CACNA1S/SCN4A) or hyperkalemic (SCN4A); episodic flaccid weakness related to K⁺ shifts
- Malignant Hyperthermia — RYR1 mutations; life-threatening hyperthermia + rigidity triggered by volatile anesthetics/succinylcholine
7. Endocrine & Toxic Myopathies
- Endocrine: Hypothyroid (elevated CK, stiffness), hyperthyroid, Cushing's syndrome (steroid myopathy — type II fiber atrophy, normal CK)
- Toxic: Statins (spectrum from myalgia → rhabdomyolysis), glucocorticoids, zidovudine (mitochondrial), alcohol, colchicine, hydroxychloroquine
8. Critical Illness Myopathy
- Occurs in ICU patients (sepsis, steroids, non-depolarizing neuromuscular blockade)
- Includes: cachectic myopathy (type II atrophy, normal CK/EMG), acute necrotizing ICU myopathy (panfascicular necrosis), thick-filament myopathy (myosin loss; associated with steroids + NMBAs)
Diagnostic Approach
| Test | Purpose |
|---|---|
| Serum CK | Most sensitive marker of muscle damage; MM isoform = skeletal muscle |
| EMG | Myopathic units (short duration, low amplitude, polyphasic); distinguishes from neuropathy |
| Nerve conduction studies | Normal in most myopathies |
| Muscle biopsy | Definitive for structural diagnosis (inflammation, fiber type, inclusions) |
| Genetic testing | Confirmatory for hereditary myopathies |
| Autoantibodies | Myositis-specific (anti-Jo-1, anti-SRP, anti-MDA5, anti-Mi-2, anti-HMGCR) |
| Forearm exercise test | No lactate rise → glycogenosis; no ammonia rise → AMP deaminase deficiency |
Key Points to Remember
- Most myopathies → proximal > distal weakness, reflexes preserved, no sensory loss
- Elevated CK points to muscle disease, but can be normal in steroid myopathy and some endocrine causes
- Aldolase can be elevated in perimysial/fascial inflammation (DM) even when CK is normal
- GGT elevation (not elevated in muscle) distinguishes liver from muscle as the source of elevated transaminases
- Myotonia = prolonged post-contraction relaxation; key in channelopathies and myotonic dystrophies
— Harrison's Principles of Internal Medicine 22e; Bradley and Daroff's Neurology in Clinical Practice
pathophysiology
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
I have comprehensive content covering all major pathophysiology categories. Let me now deliver the full synthesis.
Pathophysiology of Myopathies — Overview by Category
1. Muscular Dystrophies — Structural Membrane Failure
The central mechanism in most muscular dystrophies is disruption of the sarcolemmal membrane–cytoskeleton–extracellular matrix linkage, leading to mechanical instability and progressive fiber destruction.
The Dystrophin-Glycoprotein Complex (DGC)
Dystrophin is a large rod-shaped intracellular protein that links cytoplasmic actin (inside the myofiber) to laminin-2 in the basal lamina (outside the cell) via a chain of transmembrane proteins:
- Dystroglycans (α and β) — form the direct bridge between dystrophin and laminin
- Sarcoglycans (α, β, γ, δ) — associated transmembrane proteins that stabilize the complex
- Dystrobrevin / Syntrophins — intracellular scaffold proteins
- nNOS (neuronal nitric oxide synthase) — anchored to the complex via syntrophins; involved in vasodilatation during exercise
- Caveolin — associated with the complex
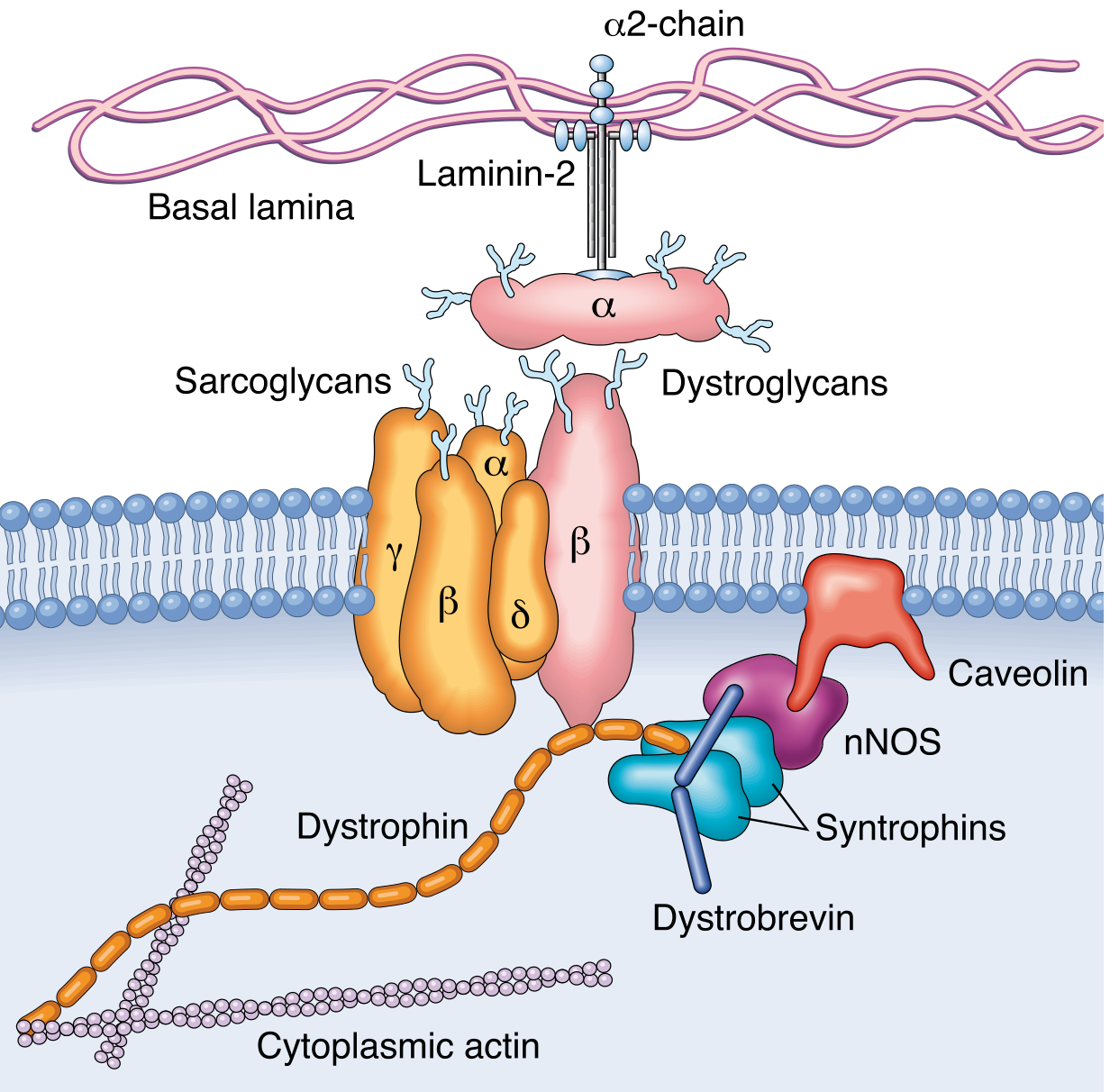
The DGC spans the sarcolemma, coupling the intracellular cytoskeleton to the extracellular matrix — Robbins Pathologic Basis of Disease
Duchenne / Becker MD (Dystrophinopathies)
- Gene: DMD on X chromosome (2.3 Mb, 79 exons — one of the largest human genes)
- DMD: Frameshift or deletion → complete absence of dystrophin
- BMD: In-frame deletions → truncated but partially functional dystrophin
Cascade of injury:
- Loss of dystrophin → DGC destabilized → sarcolemma mechanically fragile
- During contraction, micro-tears develop in the sarcolemma
- Uncontrolled Ca²⁺ influx through membrane defects
- Ca²⁺ activates proteases (calpains) and mitochondrial apoptotic pathways → myofiber necrosis
- Repeated cycles of necrosis exceed regenerative capacity (satellite cells)
- Progressive replacement by collagen and fat (fatty replacement / endomysial fibrosis)
- Loss of nNOS anchoring → impaired exercise-induced vasodilation → relative ischemia
Morphologic progression (Duchenne):
- Early: Fiber size variation, segmental necrosis, basophilic regenerating fibers, mild fibrosis
- Late: Marked atrophy + hypertrophy of remaining fibers, extensive fatty infiltration, loss of fascicular architecture
- Pseudohypertrophy of calf muscles = infiltration by fat + connective tissue, not true hypertrophy
Limb-Girdle MD (LGMDs)
- Mutations in sarcoglycans (α, β, γ, δ) — autosomal recessive forms (LGMD2C–F)
- Mutations in caveolin-3 — autosomal dominant form (LGMD1C)
- Mutations in calpain-3 (LGMD2A), dysferlin (LGMD2B) — affect membrane repair
- Common endpoint: DGC instability → same cycle of membrane damage → Ca²⁺ influx → necrosis
Emery-Dreifuss MD
- Mutations in emerin (X-linked) or lamins A/C (AD/AR) — nuclear envelope proteins
- Pathophysiology: nuclear envelope disruption → impaired mechanotransduction and nuclear integrity → muscle and cardiac muscle damage
- Distinctive early contractures (Achilles, elbow, posterior neck) and obligatory cardiac conduction defects/arrhythmias
2. Myotonic Dystrophies — RNA Toxic Gain-of-Function
Unlike structural dystrophies, myotonic dystrophies operate by an RNA gain-of-function mechanism.
- DM1 (DMPK gene, chr 19): CTG trinucleotide repeat expansion → transcribed but not translated → toxic CUG-repeat RNA accumulates in the nucleus → sequesters RNA-binding proteins (MBNL1/MBNL2) → widespread alternative splicing dysregulation of many downstream transcripts (including the chloride channel ClC-1, insulin receptor, cardiac troponin T)
- DM2 (CNBP gene, chr 3): CCTG repeat expansion → similar RNA toxicity mechanism
- Myotonia results from abnormal splicing of the CLCN1 transcript → reduced chloride channel conductance → prolonged sarcolemmal depolarization after each action potential → delayed relaxation
Channelopathies (Non-dystrophic)
- Myotonia Congenita: Loss-of-function mutations in CLCN1 (chloride channel) → same mechanism as above — reduced Cl⁻ conductance → prolonged depolarization → myotonia (without weakness)
- Paramyotonia Congenita / Hyperkalemic Periodic Paralysis: Gain-of-function mutations in SCN4A (sodium channel) → impaired channel inactivation → sustained depolarization → at mild levels produces myotonia; at severe levels produces depolarization block → flaccid paralysis
- Hypokalemic Periodic Paralysis: Mutations in CACNA1S (Ca²⁺ channel, type 1) or SCN4A → gating pore currents allowing cation leak → K⁺ shifts into cells with carbohydrate load or rest after exercise → sarcolemmal hyperpolarization → failure to generate action potentials → paralysis
3. Inflammatory Myopathies — Immune-Mediated Injury
Dermatomyositis (DM)
- Humoral / vasculopathic mechanism: Autoantibodies activate complement → C5b–9 membrane attack complex deposits on capillary endothelium → microangiopathy → dropout of intramuscular capillaries → ischemia and perifascicular microinfarcts
- Prominent type I interferon (IFN) signaling pathway activation: IFN-induced protein MxA overexpressed in muscle and endothelial cells; tubuloreticular inclusions in capillary endothelium are ultrastructural hallmarks of IFN activation
- Perifascicular atrophy — fibers at the periphery of fascicles are most ischemic and atrophy preferentially
- Myositis-specific antibodies (anti-TIF1-γ, anti-Mi-2, anti-MDA5, anti-NXP-2) associate with distinct pathologic and clinical subsets
Polymyositis / Immune-Mediated Necrotizing Myopathy (IMNM)
- Cell-mediated (CD8+ T cell) mechanism: CD8+ cytotoxic T cells invade and destroy MHC class I–expressing myofibers directly
- MHC class I is normally absent from muscle; its upregulation under inflammatory stress marks fibers for immune attack
- IMNM: Anti-SRP or anti-HMGCR antibodies → complement-mediated or antibody-dependent necrotizing injury → prominent fiber necrosis with minimal inflammatory infiltrate; statin exposure can expose HMGCR as an antigen
Inclusion Body Myositis (IBM)
- Dual pathology — both inflammatory and degenerative:
- Endomysial CD8+ T cell infiltrates invading MHC I–expressing fibers (immune component)
- Rimmed vacuoles containing aggregates of misfolded proteins: β-amyloid, TDP-43, ubiquitin, p62 — proteins shared with neurodegenerative diseases
- Filamentous inclusions on electron microscopy
- Protein homeostasis failure (impaired autophagy/ubiquitin-proteasome system) → toxic protein aggregation within myofibers
- Whether inflammation is primary or secondary to degeneration is unresolved; the end result is endomysial fibrosis and fatty replacement in a characteristic topographic pattern (finger flexors, quadriceps)
4. Metabolic Myopathies — Energy Supply Failure
Glycogen Storage Diseases (Glycogenoses)
- McArdle Disease (GSD type V): Myophosphorylase (PYGM) deficiency → glycogen cannot be broken down to glucose-1-phosphate during exercise → ATP deficit under high-demand conditions → muscle cramps, contracture, rhabdomyolysis
- "Second wind" phenomenon: After 8–10 min of aerobic exercise, free fatty acid and hepatic glucose delivery compensates, relieving symptoms
- Forearm ischemic exercise test: Lactate fails to rise (blocked glycolysis) but ammonia rises normally
- Pompe disease (GSD type II): Acid maltase (α-glucosidase) deficiency → lysosomal glycogen accumulation → lysosomal rupture → myofiber destruction; also progressive respiratory muscle failure
Lipid Metabolism Disorders
- CPT2 Deficiency: Carnitine palmitoyltransferase II deficiency → long-chain fatty acids cannot enter the mitochondrial matrix → fatty acid oxidation blocked during prolonged low-intensity exercise, fasting, or cold exposure → ATP depletion → rhabdomyolysis and myoglobinuria
- Carnitine deficiency: Impaired fatty acid transport → lipid vacuoles accumulate in myofibers
Mitochondrial Myopathies
- Mutations in mitochondrial DNA (mtDNA) or nuclear genes encoding mitochondrial proteins → defects in the electron transport chain (ETC) complexes → impaired oxidative phosphorylation → failure of aerobic ATP generation
- Heteroplasmy: mtDNA exists in multiple copies per cell; disease manifests when mutant mtDNA exceeds a threshold (threshold effect)
- Energy failure preferentially affects high-demand tissues: muscle, brain, heart, retina
- Ragged red fibers (RRF) on Gomori trichrome stain: Subsarcolemmal and intermyofibrillar accumulation of abnormal mitochondria (mitochondrial proliferation as a compensatory response to energy failure)
- Classic syndromes: MELAS (mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes), MERRF (myoclonic epilepsy with RRF), Kearns-Sayre (CPEO, pigmentary retinopathy, heart block — large mtDNA deletion)
5. Congenital Myopathies — Sarcomere Structural Defects
These arise from mutations in proteins of the contractile apparatus or its regulatory components, causing structural abnormalities visible on biopsy:
| Type | Defective Protein | Structural Lesion | Mechanism |
|---|---|---|---|
| Nemaline myopathy | Nebulin, α-tropomyosin, α-actin, troponin T | Nemaline (rod) bodies — disorganized thin filament components | Thin filament dysfunction → impaired force generation |
| Central core disease | Ryanodine receptor (RYR1) | Central areas devoid of oxidative enzymes and organelles | Abnormal Ca²⁺ release → impaired excitation-contraction coupling; also susceptibility to malignant hyperthermia |
| Centronuclear/Myotubular myopathy | Myotubularin (X-linked), dynamin-2, amphiphysin | Central nuclei instead of peripheral → arrested myofiber maturation | Defective membrane tubulation and organelle positioning |
6. Toxic Myopathies — Direct or Immune-Mediated Fiber Damage
- Statins: Inhibit HMG-CoA reductase → reduced mevalonate → depleted geranylgeranyl pyrophosphate (required for Rho GTPase prenylation and mitochondrial CoQ synthesis) → impaired mitochondrial function and membrane integrity → myofiber necrosis; in genetically predisposed individuals, can trigger IMNM via anti-HMGCR antibody generation
- Glucocorticoids: Activate proteolytic pathways (ubiquitin-proteasome, calpains) preferentially in type II (fast-twitch) fibers → selective type II atrophy; CK is typically normal (no necrosis)
- Zidovudine (AZT): Inhibits mitochondrial DNA polymerase γ → mtDNA depletion → mitochondrial dysfunction → morphologic features resembling mitochondrial myopathy (ragged red fibers)
- Alcohol: Direct membrane toxicity + mitochondrial dysfunction + hypokalemia and hypophosphatemia → acute or chronic myopathy/rhabdomyolysis
Unifying Pathologic Themes
| Mechanism | Examples |
|---|---|
| Sarcolemmal instability / Ca²⁺ influx | Duchenne/Becker, sarcoglycanopathies |
| RNA toxicity / splicing dysregulation | Myotonic dystrophy (DM1, DM2) |
| Ion channel dysfunction | Myotonia congenita, periodic paralyses, paramyotonia congenita |
| Immune-mediated fiber destruction | Dermatomyositis (vasculopathic), IBM (CD8+ T cells), IMNM |
| Energy metabolism failure | McArdle, CPT2 deficiency, mitochondrial myopathies |
| Toxic protein aggregation | Inclusion body myositis |
| Nuclear envelope disruption | Emery-Dreifuss (emerin/lamin mutations) |
| Sarcomere structural defects | Nemaline, central core, centronuclear myopathies |
— Robbins, Cotran & Kumar: Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22e; Histology: A Text and Atlas (Ross & Pawlina)
This is a shared conversation. Sign in to Orris to start your own chat.