Make easy assignment on leukemia (pathology) for pharmacy student
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive content from Robbins & Kumar Basic Pathology on leukemia. Let me compile the assignment.
📚 Leukemia — Pathology Assignment for Pharmacy Students
Course: Pathology | Level: Undergraduate Pharmacy
Source: Robbins & Kumar Basic Pathology
Learning Objectives
By the end of this assignment, students should be able to:
- Define leukemia and distinguish it from lymphoma
- Classify leukemia into its four major types
- Describe the pathogenesis, morphology, and clinical features of each type
- Identify key diagnostic markers and chromosomal abnormalities
- Relate pathophysiology to pharmacological treatment strategies
Part 1: Introduction & Classification
Leukemia is a malignant neoplasm of hematopoietic progenitor cells that accumulate primarily in the bone marrow and peripheral blood, as opposed to lymphomas, which predominantly involve lymph nodes.
Classification Overview
| Type | Cell of Origin | Course |
|---|---|---|
| AML (Acute Myeloid Leukemia) | Myeloid progenitor | Aggressive / Acute |
| ALL (Acute Lymphoblastic Leukemia) | Lymphoid progenitor | Aggressive / Acute |
| CML (Chronic Myeloid Leukemia) | Hematopoietic stem cell | Indolent / Chronic |
| CLL (Chronic Lymphocytic Leukemia) | Mature B cell | Indolent / Chronic |
Key concept: Acute leukemias = accumulation of immature blasts (arrested differentiation). Chronic leukemias = accumulation of mature but abnormal cells with uncontrolled proliferation.
Part 2: Acute Myeloid Leukemia (AML)
Pathogenesis
AML arises from mutations in genes regulating myeloid progenitor differentiation. Key mutations include:
- FLT3 (tyrosine kinase) — most common; activating mutations drive proliferation
- NPM1, CEBPA — associated with better prognosis
- t(15;17) translocation → PML-RARα fusion gene → causes Acute Promyelocytic Leukemia (APL, AML-M3)
- The fusion protein blocks granulocyte differentiation by aberrantly repressing retinoic acid target genes
Morphology
- Bone marrow replaced by myeloblasts (>20% blasts required for diagnosis)
- Blasts often contain Auer rods — needle-like cytoplasmic inclusions (pathognomonic for AML)
- Peripheral blood shows anemia, thrombocytopenia, variable WBC
Clinical Features
- Symptoms of bone marrow failure: fatigue (anemia), infections (neutropenia), bleeding (thrombocytopenia)
- Hepatosplenomegaly
- Without treatment: rapidly fatal (weeks to months)
Pharmacy Relevance
| Drug | Mechanism | Use |
|---|---|---|
| Cytarabine (ara-C) | Antimetabolite (pyrimidine analog) | Standard AML induction |
| Daunorubicin | Anthracycline / DNA intercalator | AML induction ("7+3" regimen) |
| All-trans retinoic acid (ATRA) | Differentiating agent; overcomes PML-RARα block | APL (AML-M3) — landmark therapy |
| Midostaurin | FLT3 inhibitor | FLT3-mutated AML |
| Venetoclax | BCL-2 inhibitor | Elderly/unfit AML patients |
Part 3: Acute Lymphoblastic Leukemia (ALL)
Pathogenesis
ALL arises from mutations in B-cell or T-cell progenitors. It is the most common cancer in children.
- B-ALL (~85%): often associated with translocations
- t(12;21) → TEL-AML1 — good prognosis (most common childhood ALL)
- t(9;22) → BCR-ABL (Philadelphia chromosome) — poor prognosis; ~25% of adult ALL
- T-ALL (~15%): associated with NOTCH1 mutations; mediastinal mass common in adolescent males
Morphology
- Bone marrow and blood packed with lymphoblasts
- Lymphoblasts: small to medium cells, scant cytoplasm, condensed chromatin
- No Auer rods (distinguishes from AML)
Clinical Features
- Abrupt onset: fever, pallor, bleeding, bone pain (due to marrow expansion)
- CNS involvement common → headache, cranial nerve palsies (requires CNS prophylaxis)
- Lymphadenopathy, splenomegaly
Pharmacy Relevance
| Drug | Use/Notes |
|---|---|
| Vincristine, L-asparaginase, prednisone, doxorubicin | Multi-drug induction |
| Methotrexate (intrathecal) | CNS prophylaxis |
| Imatinib / Dasatinib | BCR-ABL+ ALL (Ph+ ALL) |
| Blinatumomab | BiTE antibody (CD3×CD19); for relapsed B-ALL |
Part 4: Chronic Myeloid Leukemia (CML)
Pathogenesis
CML is defined by the Philadelphia chromosome — a balanced translocation t(9;22) creating the BCR-ABL fusion gene.
- BCR-ABL encodes a constitutively active tyrosine kinase
- It activates RAS and other proliferative pathways
- Normal myeloid progenitors require growth factors; CML progenitors are growth factor–independent
- Differentiation is preserved early → excessive but relatively normal-looking granulocytes
The discovery of BCR-ABL in CML is one of the most important milestones in oncology, as it led directly to the development of imatinib (Gleevec) — the prototype targeted cancer therapy.
Morphology
- WBC often >100,000 cells/µL
- Peripheral blood: neutrophils, immature granulocytes, basophilia and eosinophilia, elevated platelets
- Bone marrow: hypercellular with granulocytic and megakaryocytic hyperplasia
- Massive splenomegaly (extramedullary hematopoiesis → splenic infarcts)
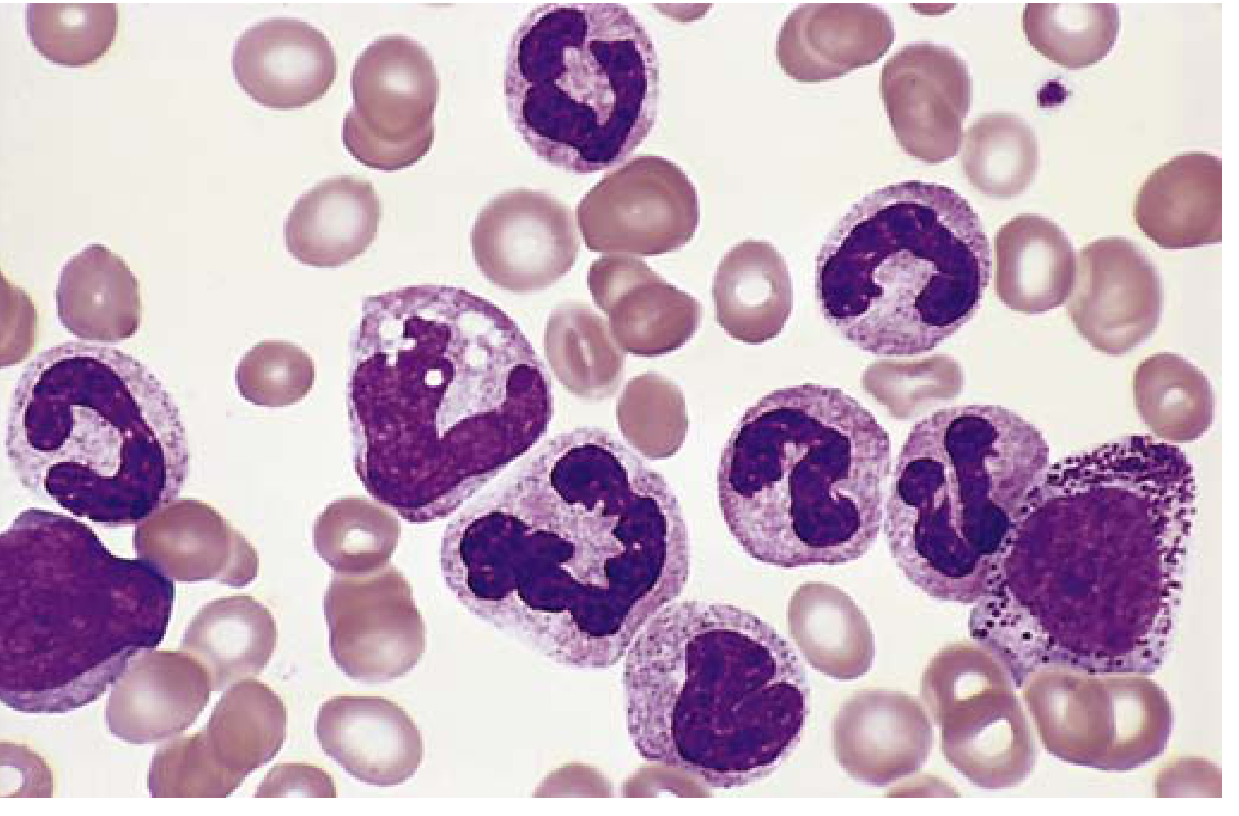
Disease Phases
| Phase | Blasts in Blood/Marrow | Features |
|---|---|---|
| Chronic | <10% | Mild symptoms, responds to therapy |
| Accelerated | 10–19% | Worsening disease |
| Blast crisis | ≥20% | Mimics acute leukemia; poor prognosis |
Clinical Features
- Insidious onset: fatigue, weight loss, night sweats
- Dragging sensation in left upper abdomen (splenomegaly)
- Labs: markedly elevated WBC with left shift, low leukocyte alkaline phosphatase (LAP) score
Pharmacy Relevance
| Drug | Mechanism | Notes |
|---|---|---|
| Imatinib (Gleevec) | BCR-ABL tyrosine kinase inhibitor (1st gen) | Revolutionized CML treatment; oral, once daily |
| Dasatinib, Nilotinib | 2nd gen TKIs | More potent; used in resistant/intolerant cases |
| Ponatinib | 3rd gen TKI | For T315I mutation ("gatekeeper" resistance) |
| Busulfan, Hydroxyurea | Cytoreductive agents | Older therapies; still used in some settings |
Part 5: Chronic Lymphocytic Leukemia (CLL)
Pathogenesis
- Neoplastic proliferation of mature but immunologically incompetent B cells
- Cells accumulate in blood, marrow, lymph nodes, and spleen
- Associated with deletions of 13q14, 11q, 17p (TP53), and trisomy 12
- 17p deletion (TP53 loss) → poor prognosis, resistance to chemotherapy
Morphology
- Blood and marrow: flood of small mature lymphocytes
- Characteristic "smudge cells" on peripheral smear (fragile lymphocytes ruptured during slide preparation)
- Lymph nodes: diffuse effacement by small round lymphocytes
Clinical Features
- Often asymptomatic, discovered incidentally on CBC
- Hypogammaglobulinemia → recurrent bacterial infections
- Autoimmune hemolytic anemia (AIHA) and thrombocytopenia in some
- Richter transformation: ~5% transform to aggressive diffuse large B-cell lymphoma
Staging (Rai System — simplified)
| Stage | Features | Risk |
|---|---|---|
| 0 | Lymphocytosis only | Low |
| I–II | + lymphadenopathy / organomegaly | Intermediate |
| III–IV | + Anemia / thrombocytopenia | High |
Pharmacy Relevance
| Drug | Mechanism | Notes |
|---|---|---|
| Ibrutinib | BTK inhibitor | 1st-line; oral targeted therapy |
| Venetoclax | BCL-2 inhibitor | Overcomes anti-apoptotic survival signal |
| Obinutuzumab / Rituximab | Anti-CD20 monoclonal antibody | Used in combination regimens |
| Fludarabine + Cyclophosphamide | Chemotherapy | Older FCR regimen for fit patients |
Part 6: Summary Comparison Table
| Feature | AML | ALL | CML | CLL |
|---|---|---|---|---|
| Peak age | Any (>50 common) | Children (2–10 yr) | 25–60 yr | >60 yr |
| Cell type | Myeloblast | Lymphoblast | Myeloid (mature) | B lymphocyte (mature) |
| Key marker | Auer rods | TdT+, CD10+ | BCR-ABL, Ph chromosome | CD5+, CD23+, smudge cells |
| Chromosome | Variable | t(12;21), t(9;22) | t(9;22) | 13q del, 17p del |
| Key drug | Cytarabine + anthracycline | Multi-agent + TKI (if Ph+) | Imatinib | Ibrutinib / Venetoclax |
| Prognosis (untreated) | Weeks | Weeks | Years | Years |
Part 7: Practice Questions
Q1. A 4-year-old child presents with pallor, bone pain, and lymphadenopathy. Bone marrow biopsy shows >90% lymphoblasts, TdT+, CD10+. What is the most likely diagnosis and first-line treatment approach?
Q2. A 55-year-old presents with fatigue and a WBC of 120,000/µL. Peripheral smear shows granulocytes at all stages of maturation with basophilia. Cytogenetics reveals t(9;22). Name the fusion gene involved and the drug of choice.
Q3. A patient with APL (AML-M3) is started on ATRA. Explain the mechanism of action at the molecular level.
Q4. Compare the peripheral blood findings in CML vs. CLL. What pathognomonic finding is seen in each?
Q5. A 70-year-old CLL patient develops worsening anemia and a rapidly enlarging lymph node mass. Biopsy shows large B cells. What has occurred, and what is this phenomenon called?
Key Terms Glossary
| Term | Definition |
|---|---|
| Blast | Immature, undifferentiated hematopoietic precursor cell |
| Auer rod | Red needle-like cytoplasmic inclusion; pathognomonic of AML |
| Philadelphia chromosome | t(9;22); creates BCR-ABL; found in CML and Ph+ ALL |
| TdT | Terminal deoxynucleotidyl transferase; marker of immature lymphoblasts |
| Smudge cell | Fragile CLL lymphocyte ruptured on peripheral smear |
| Blast crisis | Terminal phase of CML with ≥20% blasts; behaves like acute leukemia |
| Tyrosine kinase inhibitor (TKI) | Drug that blocks aberrant kinase activity (e.g., imatinib blocks BCR-ABL) |
| BTK inhibitor | Blocks B-cell receptor signaling; used in CLL (e.g., ibrutinib) |
Source: Robbins & Kumar Basic Pathology — Chapter 10: Hematopoietic and Lymphoid Systems
Make assignment on leukemia pathology for pharmacy student
Asking for Preferences
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
I now have all the core content needed from Robbins & Kumar. I have rich information on CML, and from the truncated outputs I also received detailed content on ALL, AML, and CLL. Let me now compile the full intermediate-level assignment with notes + questions + answers.
🩸 Leukemia — Pathology Assignment
For Pharmacy Students | Intermediate Level
Course: Pathology for Pharmacy | Format: Study Notes + Questions + Model Answers
Reference: Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine
SECTION A: STUDY NOTES
1. Introduction
Leukemia is a malignant clonal proliferation of hematopoietic precursor cells that primarily involves the bone marrow and peripheral blood. Unlike lymphomas, which predominantly form solid tumor masses in lymph nodes, leukemic cells flood the bloodstream and infiltrate the marrow, displacing normal hematopoiesis.
The hallmark consequence of marrow failure is the clinical triad:
- Anemia → fatigue, pallor
- Thrombocytopenia → bleeding, petechiae
- Neutropenia/leukopenia → recurrent infections
2. Classification
Leukemias are classified by:
- Lineage — Myeloid vs. Lymphoid
- Clinical course — Acute (immature blasts, rapid) vs. Chronic (mature cells, indolent)
| Type | Abbreviation | Cell Origin | Course |
|---|---|---|---|
| Acute Myeloid Leukemia | AML | Myeloid blast | Acute — weeks |
| Acute Lymphoblastic Leukemia | ALL | Lymphoid blast | Acute — weeks |
| Chronic Myeloid Leukemia | CML | Hematopoietic stem cell | Chronic — years |
| Chronic Lymphocytic Leukemia | CLL | Mature B lymphocyte | Chronic — years |
Acute leukemias = differentiation is arrested → immature blasts accumulate Chronic leukemias = differentiation is preserved → mature but abnormal cells accumulate
3. Acute Myeloid Leukemia (AML)
Epidemiology
- Most common acute leukemia in adults (median age ~65)
- ~10,000 new cases/year in the US
Pathogenesis
AML results from acquired mutations that:
- Block myeloid differentiation (class II mutations, e.g., PML-RARα, CEBPA mutations)
- Drive uncontrolled proliferation (class I mutations, e.g., FLT3, RAS activating mutations)
Key molecular subtypes:
| Mutation/Translocation | Subtype | Clinical Significance |
|---|---|---|
| t(15;17) → PML-RARα | APL (AML-M3) | Responds to ATRA + arsenic; DIC risk |
| FLT3-ITD | — | Poor prognosis; targetable with midostaurin |
| NPM1 mutation | — | Good prognosis |
| t(8;21), inv(16) | Core-binding factor AML | Good prognosis |
APL note for pharmacy students: The PML-RARα fusion protein blocks retinoic acid receptor signaling and prevents granulocyte differentiation. All-trans retinoic acid (ATRA) overcomes this block and drives terminal differentiation of blasts — a landmark example of differentiation therapy.
Morphology
- Bone marrow: >20% blasts (WHO diagnostic threshold)
- Peripheral smear: myeloblasts with large nuclei, prominent nucleoli
- Pathognomonic: Auer rods — red needle-like cytoplasmic granules in blasts; seen only in AML (not ALL)
- Extramedullary: hepatosplenomegaly; gingival hyperplasia (especially monocytic AML)
Clinical Features
- Sudden onset: fever, fatigue, bleeding, bone pain
- DIC is a serious complication, especially in APL — due to procoagulant release from granules
- CNS involvement less common than ALL
Treatment (Pharmacy Focus)
| Drug | Class | Mechanism | Use |
|---|---|---|---|
| Cytarabine (Ara-C) | Antimetabolite | Inhibits DNA polymerase (pyrimidine analog) | Backbone of AML induction |
| Daunorubicin / Idarubicin | Anthracycline | DNA intercalation + topoisomerase II inhibition | "7+3" induction regimen |
| ATRA (tretinoin) | Retinoid | Differentiating agent; overcomes PML-RARα block | APL (AML-M3) |
| Arsenic trioxide (ATO) | — | Promotes APL blast apoptosis/differentiation | APL (with ATRA) |
| Midostaurin | FLT3 inhibitor | Blocks constitutive FLT3 kinase activity | FLT3-mutated AML |
| Venetoclax | BCL-2 inhibitor | Restores apoptosis in leukemic cells | Elderly/unfit AML |
4. Acute Lymphoblastic Leukemia (ALL)
Epidemiology
- Most common cancer in children (peak age 2–10 years)
- B-ALL: ~85% of cases; T-ALL: ~15%
- In adults: Ph-chromosome positive ALL is more common and has worse prognosis
Pathogenesis
ALL arises from mutations in B- or T-lymphoid progenitors, leading to arrested maturation and uncontrolled proliferation.
Key chromosomal abnormalities:
| Translocation | Fusion Gene | Prognosis | Notes |
|---|---|---|---|
| t(12;21) | TEL-AML1 (ETV6-RUNX1) | Excellent | Most common childhood ALL |
| t(9;22) | BCR-ABL (Ph chromosome) | Poor | ~25% adult ALL; requires TKI therapy |
| t(1;19) | E2A-PBX1 | Intermediate | — |
| t(4;11) | MLL-AF4 | Very poor | Infant ALL |
Morphology
- Bone marrow: >20% lymphoblasts
- Lymphoblasts: small-to-medium, scant cytoplasm, condensed chromatin
- No Auer rods (distinguishes from AML)
- Immunophenotyping: TdT+ (terminal deoxynucleotidyl transferase — marker of lymphoblasts), CD10+ (B-ALL)
Clinical Features
- Rapid onset: fever, pallor, bone pain, lymphadenopathy, hepatosplenomegaly
- CNS involvement in ~5% at presentation → headache, vomiting, cranial nerve palsies
- Mediastinal mass: T-ALL in adolescent males (thymic involvement)
- Testicular infiltration in boys
Treatment (Pharmacy Focus)
| Drug | Role |
|---|---|
| Vincristine | Vinca alkaloid; disrupts microtubule formation; part of induction |
| L-Asparaginase | Depletes asparagine (ALL cells cannot synthesize it); unique to ALL |
| Prednisone/Dexamethasone | Lymphocytolytic corticosteroid; part of induction |
| Methotrexate (intrathecal) | CNS prophylaxis and treatment |
| Imatinib / Dasatinib | BCR-ABL TKIs; added to all Ph+ ALL regimens |
| Blinatumomab | BiTE antibody (anti-CD3 × anti-CD19); redirects T-cells to kill B-blasts |
5. Chronic Myeloid Leukemia (CML)
Epidemiology
- Peak incidence: 25–60 years; ~4,500 new cases/year in the US
Pathogenesis — The Philadelphia Chromosome
CML is defined by the t(9;22) translocation — the Philadelphia (Ph) chromosome:
Chromosome 9: ABL gene ──┐
├──► BCR-ABL fusion gene on Chr 22
Chromosome 22: BCR gene ──┘
- BCR-ABL encodes a constitutively active tyrosine kinase
- Activates downstream RAS, PI3K/AKT, and STAT5 pathways
- Reduces growth factor dependence → uncontrolled granulocyte proliferation
- Differentiation is preserved early → relatively mature-looking cells (unlike AML)
This was the first cancer linked to a specific chromosomal abnormality (Nowell & Hungerford, 1960) and the first to be cured with targeted molecular therapy (imatinib).
Morphology
- WBC often >100,000 cells/µL (markedly elevated)
- Peripheral smear: granulocytes at all stages of maturation — neutrophils, bands, metamyelocytes, myelocytes + basophilia + eosinophilia
- Elevated platelets (thrombocytosis) early
- Bone marrow: hypercellular, granulocytic and megakaryocytic hyperplasia
- Massive splenomegaly (extramedullary hematopoiesis → risk of splenic infarcts)
Disease Phases
| Phase | Blasts in Blood/Marrow | Features |
|---|---|---|
| Chronic | < 10% | Asymptomatic or mild symptoms; responds to TKIs |
| Accelerated | 10–19% | Worsening disease, resistance emerging |
| Blast crisis | ≥ 20% | Mimics acute leukemia; myeloid (70%) or lymphoid (30%); poor prognosis |
Clinical Features
- Onset insidious: fatigue, weight loss, night sweats, left upper quadrant fullness (splenomegaly)
- Low leukocyte alkaline phosphatase (LAP) score — key lab finding distinguishing CML from leukemoid reaction (where LAP is high)
- Philadelphia chromosome on cytogenetics (FISH or PCR for BCR-ABL) is diagnostic
Treatment (Pharmacy Focus)
| Drug | Generation | Notes |
|---|---|---|
| Imatinib (Gleevec) | 1st-gen TKI | Binds BCR-ABL ATP-binding site; oral; transformed CML management |
| Dasatinib, Nilotinib | 2nd-gen TKI | More potent; used in resistance or intolerance to imatinib |
| Bosutinib | 2nd-gen TKI | Alternative 2nd-line |
| Ponatinib | 3rd-gen TKI | Overcomes T315I "gatekeeper" resistance mutation |
| Hydroxyurea | Cytoreductive | Used for rapid WBC reduction; not curative |
6. Chronic Lymphocytic Leukemia (CLL)
Epidemiology
- Most common leukemia in the Western world
- Predominantly affects the elderly (median age ~65); rare before 50
- Male predominance (2:1)
Pathogenesis
CLL is a neoplasm of mature but functionally incompetent B cells (CD5+/CD23+).
Key genetic abnormalities:
| Abnormality | Frequency | Prognosis |
|---|---|---|
| del(13q14) | ~55% | Good (if sole abnormality) |
| Trisomy 12 | ~15% | Intermediate |
| del(11q) | ~15% | Poor (ATM gene loss) |
| del(17p) / TP53 loss | ~7–10% | Very poor; resistant to chemotherapy |
- CLL cells are anti-apoptotic (overexpress BCL-2), which explains their accumulation
- Immunologically defective → hypogammaglobulinemia → recurrent bacterial infections
Morphology
- Peripheral blood: massive accumulation of small, mature-looking lymphocytes
- Pathognomonic: "Smudge cells" (Gumprecht shadows) — fragile CLL lymphocytes that rupture during slide preparation
- Lymph nodes: diffuse effacement by small round lymphocytes
- Bone marrow infiltration: interstitial or nodular pattern
Staging (Rai System)
| Stage | Features | Risk |
|---|---|---|
| 0 | Lymphocytosis only | Low |
| I | + Lymphadenopathy | Intermediate |
| II | + Hepato/splenomegaly | Intermediate |
| III | + Anemia | High |
| IV | + Thrombocytopenia | High |
Clinical Features
- Often asymptomatic at diagnosis (incidental CBC finding)
- Recurrent infections (especially encapsulated bacteria — Streptococcus pneumoniae)
- Autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) in some patients
- Richter transformation (~5%): transformation to aggressive diffuse large B-cell lymphoma — sudden worsening, rapidly enlarging nodes, systemic symptoms
Treatment (Pharmacy Focus)
| Drug | Class | Mechanism | Notes |
|---|---|---|---|
| Ibrutinib | BTK inhibitor | Blocks B-cell receptor signaling → promotes apoptosis | Oral; 1st-line standard of care |
| Venetoclax | BCL-2 inhibitor | Restores apoptosis in BCL-2–overexpressing CLL cells | Often combined with obinutuzumab |
| Obinutuzumab / Rituximab | Anti-CD20 mAb | ADCC + complement-mediated lysis of B cells | Used in combination |
| Fludarabine + Cyclophosphamide + Rituximab (FCR) | Chemoimmunotherapy | Alkylation + purine analog + anti-CD20 | Older regimen; avoided in del(17p)/TP53 |
7. Master Comparison Table
| Feature | AML | ALL | CML | CLL |
|---|---|---|---|---|
| Peak age | Adults (>50) | Children (2–10 yr) | 25–60 yr | >60 yr |
| Cell of origin | Myeloid blast | Lymphoid blast | Myeloid stem cell | Mature B cell |
| Key genetic marker | Variable (FLT3, NPM1; t(15;17) in APL) | t(12;21) children; t(9;22) adults | t(9;22) BCR-ABL | del(13q), del(17p), TP53 |
| Hallmark morphology | Auer rods | TdT+; no Auer rods | WBC >100K, all-stage granulocytes, basophilia | Smudge cells |
| Splenomegaly | Mild | Mild | Massive | Moderate |
| CNS involvement | Less common | Common | Rare | Rare |
| Key drug(s) | Cytarabine + anthracycline; ATRA (APL) | Vincristine, L-asparaginase, TKIs (Ph+) | Imatinib | Ibrutinib, Venetoclax |
| Prognosis (untreated) | Weeks | Weeks | Years | Years |
SECTION B: QUESTIONS
Part I — Multiple Choice Questions (MCQs)
Q1. A 7-year-old presents with fever, bone pain, and lymphadenopathy. Bone marrow biopsy reveals >90% lymphoblasts that are TdT-positive and CD10-positive. Which translocation is most likely associated with this presentation and carries the best prognosis?
- A) t(9;22) — BCR-ABL
- B) t(15;17) — PML-RARα
- C) t(12;21) — TEL-AML1
- D) t(4;11) — MLL-AF4
Q2. A 45-year-old man has a WBC of 130,000/µL on routine labs. Peripheral smear shows granulocytes at all stages of maturation, prominent basophilia, and elevated platelets. The leukocyte alkaline phosphatase score is low. Which gene fusion is diagnostic of this condition?
- A) PML-RARα
- B) BCR-ABL
- C) TEL-AML1
- D) MLL-AF4
Q3. A patient with APL (AML-M3) is started on All-Trans Retinoic Acid (ATRA). What is the cellular mechanism of ATRA's therapeutic effect?
- A) Inhibits tyrosine kinase activity of BCR-ABL
- B) Induces DNA alkylation in leukemic blasts
- C) Overcomes differentiation block by displacing co-repressors from PML-RARα
- D) Depletes asparagine availability to leukemic blasts
Q4. Which morphological finding on peripheral blood smear is pathognomonic for AML and would NOT be seen in ALL?
- A) Smudge cells
- B) Auer rods
- C) Hypersegmented neutrophils
- D) Target cells
Q5. An elderly woman with CLL develops sudden onset of rapidly enlarging cervical lymphadenopathy, fever, and weight loss. LDH is markedly elevated. What complication has most likely occurred?
- A) Transformation to AML
- B) Autoimmune hemolytic anemia
- C) Richter transformation to diffuse large B-cell lymphoma
- D) Splenic infarction
Q6. A CML patient on imatinib develops resistance. Molecular testing reveals a T315I mutation in BCR-ABL. Which drug would be appropriate?
- A) Dasatinib
- B) Nilotinib
- C) Ponatinib
- D) Bosutinib
Q7. Which of the following best explains why CLL cells accumulate in large numbers despite being mature, non-dividing cells?
- A) Rapid cell division driven by BCR-ABL kinase
- B) Overexpression of BCL-2 → resistance to apoptosis
- C) FLT3 mutation causing growth factor independence
- D) Loss of TP53 causing uncontrolled proliferation
Q8. L-Asparaginase is a unique drug used specifically in ALL treatment. Its mechanism is:
- A) Inhibiting topoisomerase II in lymphoblasts
- B) Depleting serum asparagine, which ALL cells cannot synthesize independently
- C) Binding to CD20 on B-lymphoblasts
- D) Blocking folate metabolism in dividing lymphoblasts
Part II — Short Answer Questions (SAQs)
Q9. Explain the significance of the Philadelphia chromosome in two different leukemias. How does its presence affect treatment?
Q10. Compare and contrast the peripheral blood findings of CML and CLL, including any pathognomonic findings for each.
Q11. A pharmacy student reads that venetoclax is used in both AML and CLL. Explain the common molecular rationale for using a BCL-2 inhibitor in both diseases.
Q12. What is the "7+3" induction regimen for AML? Name the drugs and their mechanisms of action.
SECTION C: MODEL ANSWERS
MCQ Answers
| Q | Answer | Rationale |
|---|---|---|
| 1 | C | t(12;21)/TEL-AML1 is the most common childhood ALL translocation and carries excellent prognosis (~90% cure rate) |
| 2 | B | BCR-ABL from t(9;22) defines CML. Low LAP score is a hallmark distinguishing CML from reactive leukocytosis |
| 3 | C | PML-RARα acts as a dominant repressor. ATRA binds RARα, displaces co-repressor complexes, and restores normal transcription needed for granulocyte differentiation |
| 4 | B | Auer rods are crystallized azurophilic granules seen only in AML myeloblasts — never in ALL |
| 5 | C | Richter transformation = CLL → aggressive DLBCL; marked by sudden lymph node enlargement, B symptoms, high LDH |
| 6 | C | T315I is the "gatekeeper" resistance mutation; it blocks binding of all 1st and 2nd generation TKIs except ponatinib (3rd gen) |
| 7 | B | CLL cells overexpress BCL-2 (an anti-apoptotic protein), preventing programmed cell death → accumulation of long-lived cells |
| 8 | B | Normal cells synthesize asparagine; ALL cells lack asparagine synthetase and depend on serum supply. L-Asparaginase depletes serum asparagine, starving leukemic blasts |
SAQ Model Answers
Q9 — Philadelphia Chromosome in CML vs. ALL
The Philadelphia chromosome is a balanced translocation t(9;22) resulting in the BCR-ABL fusion gene, which encodes a constitutively active tyrosine kinase. It is the defining genetic lesion of CML (present in ~95% of cases) and is also found in ~25% of adult ALL (Ph+ ALL).
In CML: BCR-ABL (p210 isoform) drives myeloid stem cell proliferation with preserved differentiation. Treatment with imatinib (a BCR-ABL TKI) has transformed CML from a fatal disease to a manageable chronic condition; most patients achieve molecular remission and near-normal life expectancy.
In Ph+ ALL: BCR-ABL (p190 isoform, more common) drives lymphoblast proliferation with arrested differentiation. Presence of Ph chromosome in ALL historically indicated very poor prognosis, but adding TKIs (dasatinib or imatinib) to chemotherapy significantly improves outcomes.
Q10 — Peripheral Blood: CML vs. CLL
| Feature | CML | CLL |
|---|---|---|
| WBC | Very high (often >100,000/µL) | Elevated (mature lymphocytes) |
| Cell type | Granulocytes at all maturation stages | Small mature lymphocytes |
| Basophilia | Prominent — characteristic finding | Absent |
| Thrombocytosis | Common early | Thrombocytopenia late |
| Pathognomonic finding | All-stage granulocytes + basophilia; low LAP score | Smudge cells (ruptured fragile lymphocytes) |
In CML, differentiation is preserved so the blood looks like a "bone marrow in the blood." In CLL, the cells are morphologically mature but functionally incompetent.
Q11 — Venetoclax in AML and CLL: Common Molecular Rationale
BCL-2 is an anti-apoptotic protein located on the mitochondrial outer membrane. It sequesters pro-apoptotic proteins (BAX, BAK), preventing release of cytochrome c and caspase activation.
- In CLL: CLL cells constitutively overexpress BCL-2 as a survival mechanism — this is why they accumulate (they don't die). Venetoclax displaces pro-apoptotic proteins from BCL-2, restoring apoptosis.
- In AML: AML blasts (especially in elderly patients with lower-intensity mutations) also rely heavily on BCL-2 for survival, particularly in the setting of low oxidative phosphorylation dependency.
Thus, venetoclax exploits the BCL-2 dependency shared by both CLL and certain AML subtypes, and is a prime example of rational, mechanism-based pharmacology — targeting a shared survival pathway rather than the leukemia-specific driver mutation.
Q12 — "7+3" Induction Regimen for AML
The standard induction for AML:
- Cytarabine (Ara-C) × 7 days by continuous IV infusion
- Mechanism: Pyrimidine analog; incorporated into DNA → inhibits DNA polymerase → chain termination; S-phase specific
- Daunorubicin (or Idarubicin) × 3 days IV bolus
- Mechanism: Anthracycline; intercalates DNA → inhibits topoisomerase II → double-strand breaks; also generates free radicals
This "7+3" combination achieves complete remission in ~60–80% of younger patients. The goal is to eliminate the bulk leukemic burden and allow normal marrow recovery, followed by consolidation therapy.
Glossary of Key Terms
| Term | Definition |
|---|---|
| Blast | Immature, undifferentiated hematopoietic cell; >20% in marrow = acute leukemia |
| Auer rod | Pathognomonic needle-like cytoplasmic inclusion in AML myeloblasts |
| Philadelphia chromosome | t(9;22); creates BCR-ABL fusion gene; in CML and Ph+ ALL |
| TdT | Terminal deoxynucleotidyl transferase; nuclear enzyme marking immature lymphoblasts |
| Smudge cell | Fragile CLL lymphocyte ruptured during peripheral smear preparation |
| Blast crisis | CML terminal phase (≥20% blasts); behaves like acute leukemia |
| TKI | Tyrosine kinase inhibitor (e.g., imatinib); blocks aberrant kinase signals |
| BCL-2 | Anti-apoptotic protein; overexpressed in CLL; targeted by venetoclax |
| BTK | Bruton's tyrosine kinase; mediates B-cell receptor survival signals; inhibited by ibrutinib |
| Richter transformation | CLL → aggressive DLBCL; poor prognosis |
| LAP score | Leukocyte alkaline phosphatase; low in CML, high in leukemoid reaction |
| ATRA | All-trans retinoic acid; differentiation therapy for APL (AML-M3) |
References: Robbins & Kumar Basic Pathology (10th ed.), Chapter 10; Goldman-Cecil Medicine (27th ed.), Chapter 168.
This is a shared conversation. Sign in to Orris to start your own chat.