Difference between nephritis and nephrotic syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Here is a thorough comparison:
Nephritis vs. Nephrotic Syndrome
These two terms describe distinct clinical syndromes of glomerular disease, differing in their underlying pathophysiology, presenting features, and causes.
Core Pathophysiology
| Nephrotic Syndrome | Nephritic Syndrome (Nephritis) | |
|---|---|---|
| Primary defect | Increased glomerular capillary wall permeability to protein | Glomerular inflammation with injury to capillary walls |
| Main consequence | Protein loss in urine | Blood leaks into urine; GFR falls |
In nephrotic syndrome, the barrier function of the glomerulus is disrupted — proteins (especially albumin) pour into the filtrate. In nephritic syndrome, the glomerulus is inflamed — leukocytes infiltrate, cells proliferate, and capillary walls are damaged enough to let red blood cells through. — Comprehensive Clinical Nephrology, 7th Ed. / Robbins Pathologic Basis of Disease
Clinical Features Compared
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Onset | Insidious | Abrupt |
| Proteinuria | >3.5 g/day (massive, ++++) | Mild–moderate (non-nephrotic range, ++) |
| Hematuria | Absent or mild | +++ (hallmark) — dysmorphic RBCs, RBC casts |
| Edema | Severe (++++) — periorbital, peripheral, ascites, anasarca | Mild–moderate (++) |
| Hypertension | Normal or mildly raised | Raised (sodium retention + renin release) |
| Jugular venous pressure | Normal/low | Raised |
| Serum albumin | Low (hypoalbuminemia) | Normal or slightly reduced |
| Serum lipids | Hyperlipidemia | Usually normal |
| GFR / Renal function | Usually preserved (except some causes) | Reduced — oliguria, azotemia |
| RBC casts in urine | Absent | Present |
— Comprehensive Clinical Nephrology, 7th Edition, TABLE 16.4
Nephrotic Syndrome — Details
Defining criteria: Proteinuria >3.5 g/day/1.73 m², hypoalbuminemia, edema, hyperlipidemia (and lipiduria).
Pathophysiology of complications:
- Heavy protein loss → hypoalbuminemia → reduced oncotic pressure → edema
- Liver compensates by upregulating lipoprotein synthesis → hyperlipidemia → lipiduria
- Loss of immunoglobulins → susceptibility to infection
- Loss of antithrombin III and protein C/S → hypercoagulability (especially renal vein thrombosis)
Primary causes (in decreasing prevalence):
- Focal segmental glomerulosclerosis (FSGS)
- Membranous nephropathy (associated with anti-PLA2R antibodies)
- Minimal change disease (MCD)
- Membranoproliferative glomerulonephritis (MPGN)
Secondary causes: Diabetic nephropathy, amyloidosis, membranous lupus nephritis, HIV-associated nephropathy
— Goldman-Cecil Medicine
Nephritic Syndrome — Details
Defining criteria: Glomerular hematuria (dysmorphic RBCs or RBC casts) + hypertension + edema + reduced GFR ± oliguria + non-nephrotic proteinuria.
Pathophysiology: Proliferation of glomerular cells + leukocyte infiltration → capillary wall injury → RBCs enter urine + hemodynamic changes → reduced GFR → fluid retention → azotemia + hypertension (from fluid overload and renin release from ischemic kidneys).
Causes:
- ANCA-associated vasculitis (granulomatosis with polyangiitis, microscopic polyangiitis) — rapidly progressive course
- Anti-GBM disease (Goodpasture syndrome)
- Postinfectious GN (e.g., post-streptococcal — most classic, due to immune complex deposition)
- Immune complex GN: SLE (lupus nephritis), cryoglobulinemia, IgA nephropathy, IgA vasculitis (HSP), endocarditis
— Goldman-Cecil Medicine / Robbins Pathologic Basis of Disease
Memory Aid
| Nephrotic | Nephritic | |
|---|---|---|
| Key word | Oedema, prOtein | Inflammation, blood |
| Urine | Frothy (protein) | Smoky/cola-colored (blood) |
| Classic example | Minimal change disease (children) | Post-strep GN (children) |
Overlap
Some diseases can present with features of both syndromes (e.g., membranoproliferative GN, lupus nephritis class III/IV). Rapidly progressive glomerulonephritis (RPGN / crescentic GN) is the most severe form of the nephritic syndrome and can progress to end-stage renal disease within weeks if untreated.
Minimal change disease (children) Whats this?
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Minimal Change Disease (MCD)
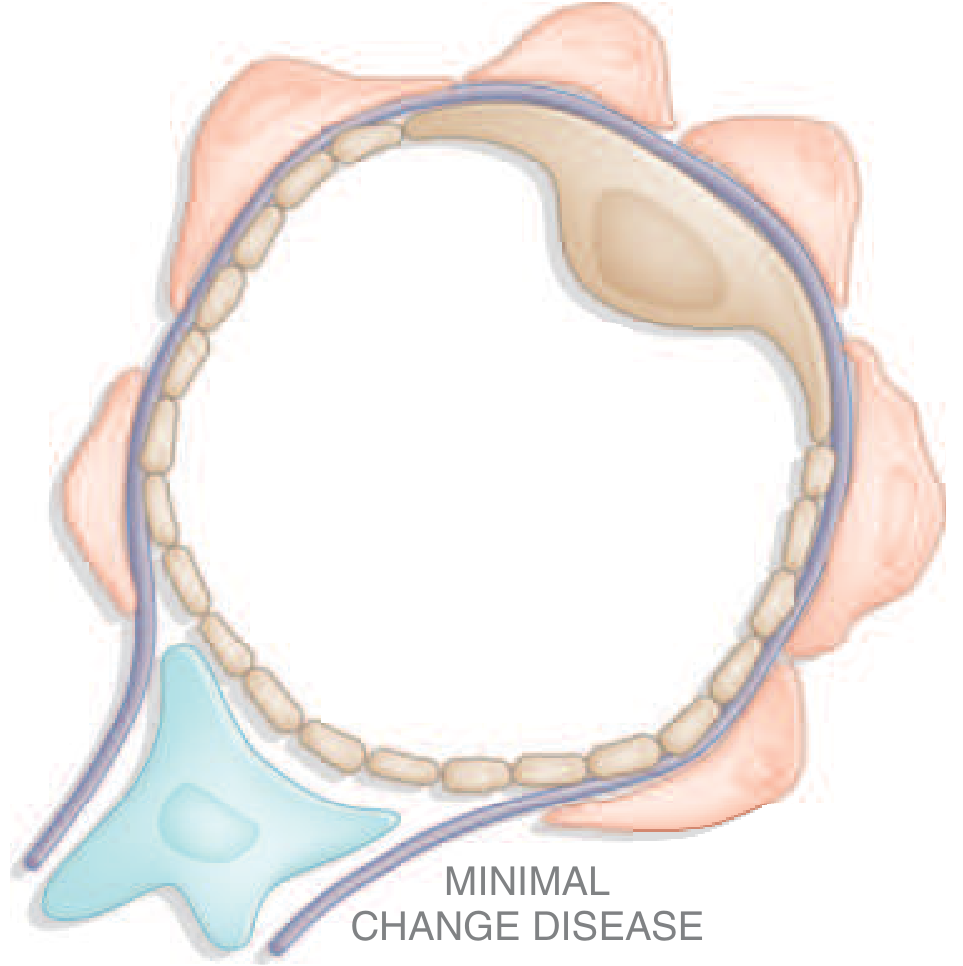
Minimal Change Disease is the most common cause of nephrotic syndrome in children, accounting for 70–90% of cases in childhood. The peak age is 2–6 years. It can also occur in adults, but is less common there (10–15%).
Why "Minimal Change"?
The name comes from the fact that the glomeruli look completely normal under light microscopy — there's "minimal change" visible. The problem is only detectable under electron microscopy (EM), which reveals the hallmark lesion.
The Key Lesion: Podocyte Foot Process Effacement
The glomerular filter has specialized cells called podocytes, which wrap around the capillary with tiny finger-like projections called foot processes. Between these processes are tiny gaps called slit diaphragms — the main filter for protein.
In MCD, the foot processes are diffusely effaced (flattened/retracted) — they lose their architecture and the slit diaphragms collapse. This makes the filter "leaky," allowing massive amounts of protein (especially albumin) to spill into the urine.
| Microscopy | Finding |
|---|---|
| Light microscopy | Normal |
| Immunofluorescence | Negative (no immune deposits) |
| Electron microscopy | Diffuse foot process effacement (the diagnostic finding) |
— Robbins, Cotran & Kumar Pathologic Basis of Disease
Pathogenesis
The exact cause is not fully understood, but immune dysfunction is the key trigger:
- T-cell dysfunction — activated T cells may secrete circulating permeability factors (cytokines like IL-13, IL-4) that directly injure podocytes
- Anti-nephrin antibodies — a subset of patients have autoantibodies against nephrin, a key slit diaphragm protein, which disrupts the filter
- CD80 (B7.1) upregulation on podocytes — disrupts normal T-cell regulation
- Loss of glomerular polyanions (charge barrier) also contributes to proteinuria
The disease sometimes follows a respiratory infection or routine immunization, supporting an immune trigger.
— Harrison's Principles of Internal Medicine 22E / Robbins Pathologic Basis of Disease
Clinical Presentation
- Abrupt onset of massive edema (periorbital, peripheral, ascites)
- Heavy proteinuria — average ~10 g/24 hr; predominantly selective (albumin only, no large proteins)
- Severe hypoalbuminemia
- No hematuria (urine sediment is acellular — "quiet urine")
- No hypertension (usually)
- Normal renal function in most cases
- Hyperlipidemia and lipiduria (typical of nephrotic syndrome)
Treatment & Prognosis — Excellent
The hallmark of MCD is its dramatic response to corticosteroids:
| Response Group | Details |
|---|---|
| Primary responders | Complete remission after one course of prednisone — >90% of children |
| Steroid-dependent | Relapse when steroids are tapered |
| Frequent relapsers | ≥2 relapses in 6 months after taper |
| Steroid-resistant | Fail to respond (may actually have FSGS on deeper biopsy) |
- 70–75% of children relapse after first remission, but relapse frequency decreases after puberty
- Long-term prognosis is excellent — even steroid-dependent disease often resolves by puberty
- For refractory cases: rituximab (anti-CD20), cyclophosphamide, or cyclosporine are used
Why Children Are Usually NOT Biopsied
Because MCD is so overwhelmingly common in young children with nephrotic syndrome (and responds so reliably to steroids), a kidney biopsy is not done first — treatment with steroids is started empirically. Biopsy is reserved for non-responders, atypical presentations, or suspected FSGS.
— Harrison's Principles of Internal Medicine 22E / Comprehensive Clinical Nephrology, 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.