Teach me thalassemia include pathogenesis, morphology, laboratory diagnosis, complications, and treatment In simple way
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive, well-illustrated answer. Let me compose the full response.
Thalassemia - A Complete Guide
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Tietz Textbook of Laboratory Medicine; Goldman-Cecil Medicine
What Is Thalassemia?
Thalassemia is a group of inherited blood disorders in which a person's body makes too little hemoglobin (Hb) - the protein in red blood cells that carries oxygen. The name comes from the Greek thalassa ("sea"), because it was first described in children living around the Mediterranean Sea.
The root cause: mutations in the genes that code for globin chains (the building blocks of hemoglobin). Adult hemoglobin (HbA) is made of 2 alpha (α) chains + 2 beta (β) chains. When one type is underproduced, the other chain accumulates and causes damage.
1. PATHOGENESIS (How the Disease Happens)
The Core Problem
Normal hemoglobin = 2 α-chains + 2 β-chains working in balance. In thalassemia, one side of that pair is deficient. The excess chain accumulates and precipitates, causing all the downstream damage.
β-Thalassemia (most clinically important)
The β-globin gene is on chromosome 11. Over 100 different point mutations can cause disease. They fall into three types:
| Mutation Type | Effect | Result |
|---|---|---|
| Splicing mutations | Abnormal mRNA splicing | β⁺ (most common) |
| Promoter mutations | Reduced transcription by 75-80% | β⁺ |
| Chain terminator mutations | Premature stop codon / frameshift | β⁰ (most severe) |
- β⁰ = NO β-globin produced
- β⁺ = REDUCED β-globin produced
How β-thalassemia causes anemia (two mechanisms):
- Hemoglobin deficiency - fewer β-chains = less HbA = pale, small, poorly oxygenated red cells (hypochromic, microcytic)
- Unpaired α-chain toxicity - excess α-chains precipitate inside red cell precursors, causing membrane damage → apoptosis of up to 70-85% of red cell precursors in severe disease ("ineffective erythropoiesis")
Downstream consequences of ineffective erythropoiesis:
- Marrow expansion (up to 30-fold) - erodes bones, causes skeletal deformities
- Extramedullary hematopoiesis - the liver, spleen, lymph nodes, and even thorax start making blood cells
- Iron overload - erythroid precursors release erythroferrone → suppresses hepcidin → excessive gut iron absorption; worsened by repeated transfusions → secondary hemochromatosis
α-Thalassemia
The 2 α-globin genes are on chromosome 16 (4 genes total). This type is mostly caused by gene deletions (not point mutations). The severity scales with how many genes are deleted:
| Genes Deleted | Syndrome | Outcome |
|---|---|---|
| 1 gene (-/αα) | Silent carrier | No symptoms |
| 2 genes (--/αα or -α/-α) | α-Thalassemia trait | Mild anemia |
| 3 genes (--/-α) | HbH disease | Moderate-severe anemia; β-chain tetramers (HbH) form |
| 4 genes (--/--) | Hydrops fetalis | Lethal in utero (γ-chain tetramers = Hb Bart's) |
2. MORPHOLOGY (What You See Under the Microscope and at Autopsy)
Peripheral Blood Smear - β-Thalassemia Major
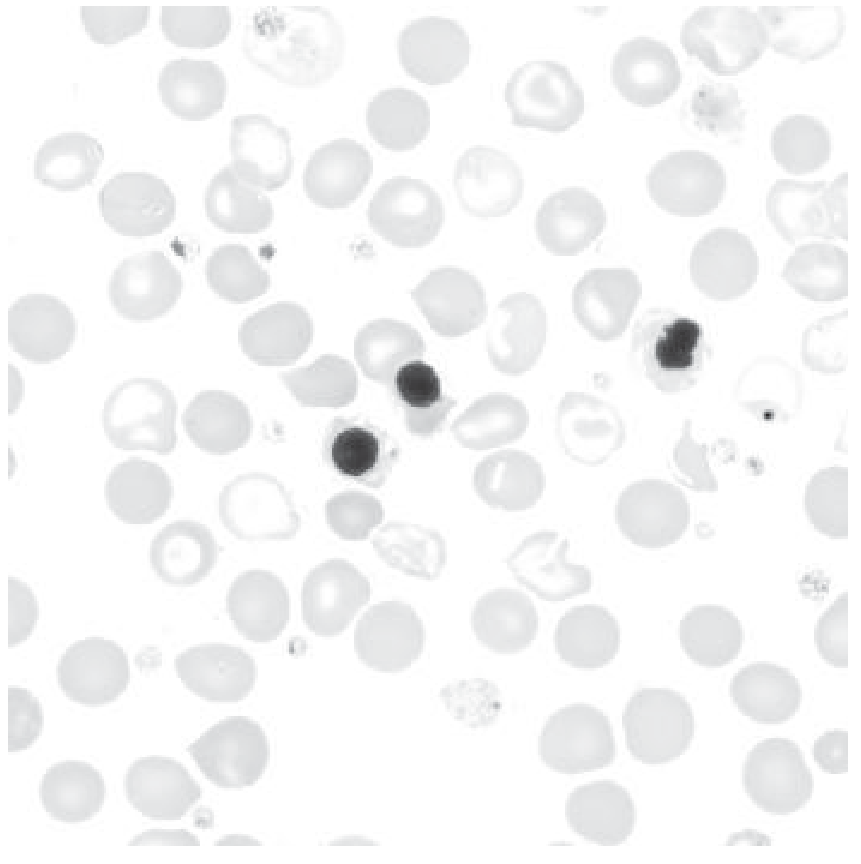
Peripheral blood smear in β-thalassemia major - note the pale, small red cells, target cells, and nucleated red blood cells
What you see on smear:
- Microcytes (small cells) and variable macrocytes
- Target cells (cells that look like a bullseye) - characteristic
- Hypochromia (pale cells with too little Hb)
- Basophilic stippling (RNA remnants in cytoplasm)
- Nucleated red blood cells (NRBCs) - a hallmark; immature cells released from marrow
- Polychromasia (blue-tinged young RBCs)
- Occasional schistocytes and spherocytes
Bone Marrow
- Marked erythroid hyperplasia (marrow is packed with red cell precursors trying to compensate)
- Many precursors dying before they mature (ineffective erythropoiesis)
Organs
- Splenomegaly - enlarged spleen from extramedullary hematopoiesis + trapping of abnormal RBCs
- Hepatomegaly - same reason
- Skull X-ray: classic "hair on end" or "sun-ray" appearance from cortical bone expansion
- Facial bones: frontal bossing, prominent cheekbones (chipmunk facies) - from marrow expansion
- Heart: iron deposits in myocardium (cardiac siderosis) in advanced cases
- Liver: iron deposition → cirrhosis in severe/untreated cases
3. LABORATORY DIAGNOSIS
Complete Blood Count (CBC)
| Parameter | Finding |
|---|---|
| Hemoglobin | Severely low (30-65 g/L in thalassemia major) |
| MCV | Low: 48-72 fL (microcytic) |
| MCH | Low: 23-32 pg |
| RBC count | Often elevated (many small, pale cells) |
| WBC + Platelets | Usually normal |
Peripheral Blood Smear
- Microcytes, target cells, NRBCs, basophilic stippling, hypochromia (as described above)
Hemoglobin HPLC / Electrophoresis (most important test)
This separates different types of hemoglobin and is the gold standard:
| Type | HbA | HbA2 | HbF |
|---|---|---|---|
| Normal | ~97% | ~2.5% | <1% |
| β-Thal minor | ↓ | ↑ >3.5% (key finding) | slightly ↑ |
| β-Thal major | Absent (β⁰) or very low | Variable | Dominant (major peak) |
| HbH disease | ↓ | ↓ | Variable; HbH peak present |
Key points:
- ↑ HbA2 (>3.5%) is the diagnostic hallmark of β-thalassemia trait/minor
- Absence of HbA with dominant HbF = β-thalassemia major
- HbH inclusions seen on brilliant cresyl blue stain in α-thalassemia (HbH disease)
Other Lab Findings
- Serum ferritin / iron studies: normal or elevated (never low - distinguishes from iron deficiency anemia!)
- Bilirubin: elevated (unconjugated) - from hemolysis
- Urobilinogen: elevated in urine
- Reticulocyte count: elevated (bone marrow trying to compensate)
- LDH: elevated (marker of cell destruction)
- Coombs test: NEGATIVE (helps distinguish from autoimmune hemolytic anemia)
- DNA analysis: definitive diagnosis, identifies specific mutations; required for genetic counseling
Key distinction from iron deficiency anemia: Both give microcytic, hypochromic anemia, but in thalassemia the serum iron/ferritin is normal or high, HbA2 is elevated, and the RBC count is typically higher relative to the Hb level.
4. COMPLICATIONS
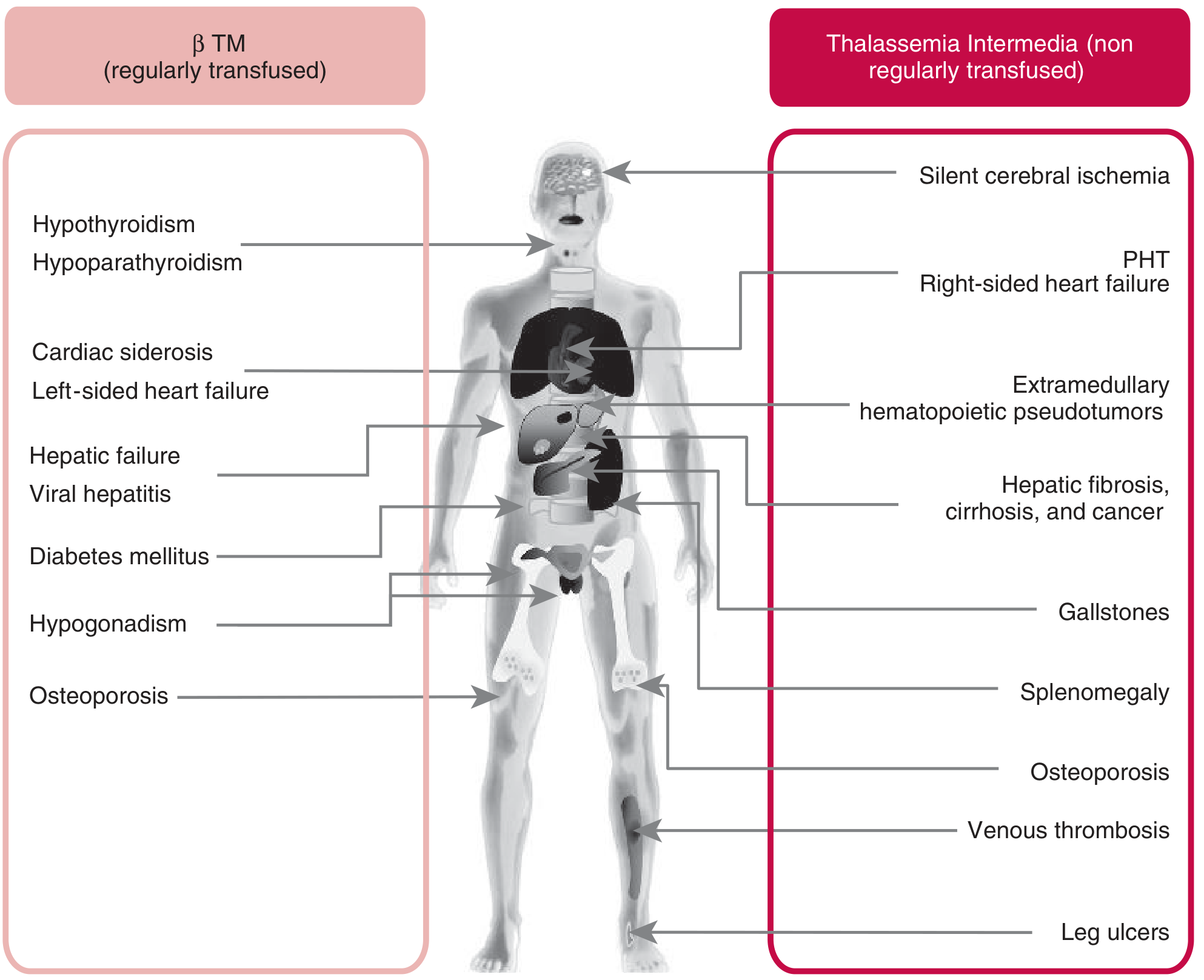
Clinical complications in β-thalassemia major (regularly transfused, left) vs. thalassemia intermedia (non-regularly transfused, right)
Complications come from two sources: the disease itself and treatment (transfusions + iron overload):
From the Disease
| System | Complication |
|---|---|
| Bone | Osteoporosis, fractures, skeletal deformities (frontal bossing, chipmunk face), "hair on end" skull |
| Spleen | Massive splenomegaly → hypersplenism (destroys blood cells + platelets) |
| Growth | Failure to thrive, short stature, delayed puberty |
| Cardiovascular | Pulmonary hypertension, right-sided heart failure (especially in intermedia) |
| Gallbladder | Gallstones (from chronic hemolysis → bilirubin stones) |
| Liver | Hepatomegaly, eventually hepatic fibrosis/cirrhosis |
| Neurological | Silent cerebral ischemia (especially in thalassemia intermedia) |
| Legs | Leg ulcers (in intermedia/non-transfused patients) |
From Treatment (Iron Overload - most dangerous)
Every 2-3 units of blood adds ~200-250 mg of iron. The body has no way to excrete excess iron, so it deposits in organs:
| Organ Affected | Consequence |
|---|---|
| Heart | Cardiac siderosis → arrhythmias, cardiac failure (leading cause of death) |
| Liver | Iron deposition → cirrhosis, hepatocellular carcinoma |
| Pancreas | Damage to beta cells → diabetes mellitus |
| Endocrine glands | Hypogonadism (no puberty), hypothyroidism, hypoparathyroidism, adrenal insufficiency |
| Pituitary | Hypogonadotropic hypogonadism → infertility |
From Blood Transfusions
- Infections: Hepatitis B, Hepatitis C, HIV (in poorly screened blood supplies)
- Alloimmunization: Immune reactions to donor red cell antigens
5. TREATMENT
A. Blood Transfusion (cornerstone of management)
- Regular transfusions every 2-5 weeks
- Goal: maintain pre-transfusion Hb above 9-10.5 g/dL
- Use leukoreduced packed red cells to minimize reactions and infection transmission
- Suppresses the body's own ineffective erythropoiesis (reduces bone changes, extramedullary hematopoiesis)
B. Iron Chelation (essential with transfusion therapy)
Because transfusions cause iron overload, chelation therapy must be given alongside:
| Drug | Route | Notes |
|---|---|---|
| Deferoxamine (Desferal) | Subcutaneous/IV infusion | Traditional gold standard; inconvenient (8-12hr infusion) |
| Deferiprone | Oral | Good cardiac iron clearance; approved even in young children |
| Deferasirox | Oral | Once daily; convenient; commonly used now |
- Start iron chelation early to protect the heart, liver, and endocrine glands
- Monitor serum ferritin and cardiac T2* MRI to guide therapy
C. Splenectomy
- Considered when hypersplenism significantly increases blood requirements (increasing iron loading despite good chelation)
- Major risk: overwhelming post-splenectomy infection (mortality ~50%) - requires vaccinations (pneumococcal, meningococcal, Hib) and prophylactic penicillin
- Delay until at least age 5 if possible
D. Hydroxyurea
- Stimulates production of fetal hemoglobin (HbF), which can partially compensate
- More useful in thalassemia intermedia and some cases of HbH disease
- Reduces transfusion requirements in some patients
E. Hematopoietic Stem Cell Transplantation (HSCT)
- The only curative treatment currently available
- Best results in young patients with HLA-matched sibling donor, before organ damage occurs
- Limited by donor availability and transplant-related risks
F. Gene Therapy (emerging)
- Betibeglogene autotemcel (Zynteglo): FDA-approved gene therapy for transfusion-dependent β-thalassemia
- Patients receive a modified copy of the β-globin gene via lentiviral vector
- Many patients become transfusion-independent
- Exciting but costly and limited in availability
G. New Agents
- Luspatercept: an activin receptor ligand trap that reduces ineffective erythropoiesis; reduces transfusion burden in transfusion-dependent β-thalassemia
- Sotatercept: similar mechanism, under investigation
Quick Summary Table
| Feature | β-Thalassemia Major | β-Thalassemia Minor | α-Thalassemia (HbH) |
|---|---|---|---|
| Genetics | Homozygous (β⁰/β⁰ or β⁰/β⁺) | Heterozygous (β/β⁰ or β/β⁺) | 3 α-gene deletions |
| Severity | Severe (transfusion-dependent) | Mild (usually asymptomatic) | Moderate |
| HbA2 | Variable | >3.5% (diagnostic) | Low |
| HbF | Dominant | Slight ↑ | Normal-slight ↑ |
| HbH | Absent | Absent | Present |
| Treatment | Transfusion + chelation | None needed | Variable |
The Big Picture in One Sentence
In thalassemia, the imbalance between α- and β-globin chains is the engine of all disease: the excess chain precipitates, destroys red cell precursors in the bone marrow (ineffective erythropoiesis), which drives bone changes, organ enlargement, iron overload, and ultimately multi-organ failure if untreated.
This is a shared conversation. Sign in to Orris to start your own chat.