Typical manifestation of cardiovascular system dysfunction . Compensation mechanism..
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I now have rich content from Braunwald's Heart Disease. Let me compile this into a comprehensive answer drawing on the retrieved content and core cardiovascular pathophysiology knowledge.
Typical Manifestations of Cardiovascular System Dysfunction & Compensation Mechanisms
I. TYPICAL MANIFESTATIONS OF CARDIOVASCULAR DYSFUNCTION
Cardiovascular dysfunction produces a spectrum of clinical signs and symptoms depending on whether the left heart, right heart, or both are involved, and whether dysfunction is systolic or diastolic.
A. Symptoms
| Symptom | Mechanism |
|---|---|
| Dyspnea (exertional → at rest → orthopnea → PND) | Elevated left ventricular end-diastolic pressure → pulmonary venous congestion → pulmonary edema |
| Fatigue & exercise intolerance | Reduced cardiac output → skeletal muscle hypoperfusion |
| Peripheral edema | Right-sided congestion → elevated systemic venous pressure → fluid extravasation |
| Nocturia | Redistribution of interstitial fluid to the circulation in the supine position → increased renal perfusion at night |
| Ascites / hepatomegaly | Elevated right atrial pressure → systemic venous hypertension |
| Chest pain / angina | Myocardial ischemia from mismatch between oxygen supply and demand |
| Palpitations / syncope | Arrhythmias (AF, VT) secondary to myocardial remodeling or ischemia |
| Cognitive impairment | Reduced cerebral perfusion |
B. Signs
- Raised JVP — reflects elevated right-sided filling pressures
- Displaced apex beat / S3 gallop — ventricular dilatation and rapid ventricular filling
- Pulmonary crackles — transudation of fluid into alveoli
- Pleural effusion — elevated pulmonary capillary pressure or systemic venous hypertension
- Cardiomegaly on CXR — ventricular remodeling
- Cool extremities & delayed capillary refill — reduced peripheral perfusion
- Pulsus alternans — marker of severe LV dysfunction
C. Congestion vs. Low Output
Most HF presentations involve congestion (elevated filling pressures) as the dominant pathway:
"Systemic or pulmonary congestion, often due to a high ventricular diastolic pressure, dominates the clinical presentation of most patients hospitalized for acute HF." — Braunwald's Heart Disease
A subset presents with low-output (cool, hypoperfused): hypotension, oliguria, altered mentation — often termed cardiogenic shock.
II. COMPENSATION MECHANISMS
When the heart is injured or stressed, the body activates several short-term adaptive mechanisms. While initially beneficial, these become maladaptive in chronic disease.
1. Frank-Starling Mechanism
- Increased venous return → ventricular dilatation → increased end-diastolic volume → greater stretch of sarcomeres → increased stroke volume
- Short-term benefit: maintains cardiac output
- Chronic problem: excessive dilatation → wall stress increases → further dysfunction (Laplace's law: wall stress = P × r / 2h)
2. Neurohormonal Activation
The dominant early compensatory response:
a. Sympathetic Nervous System (SNS)
- Increased catecholamines (norepinephrine) → tachycardia + increased contractility + vasoconstriction
- Maintains perfusion pressure to vital organs
- Chronic effects: receptor downregulation, myocyte apoptosis, arrhythmias, further myocardial damage
b. Renin-Angiotensin-Aldosterone System (RAAS)
- Reduced renal perfusion → renin release → angiotensin II → vasoconstriction + aldosterone → sodium and water retention → increased preload
- Chronic effects: myocardial fibrosis, ventricular remodeling, worsening congestion
c. Antidiuretic Hormone (ADH / Vasopressin)
- Released in response to low cardiac output and high angiotensin II → water retention → maintains circulating volume
- Contributes to dilutional hyponatremia in advanced HF
d. Natriuretic Peptides (BNP, ANP) — Counter-regulatory
- Released by stretched atria and ventricles → vasodilation + natriuresis + diuresis
- Oppose RAAS and SNS; basis for BNP as a diagnostic/prognostic marker
3. Ventricular Remodeling
Long-term structural change in response to wall stress:
| Type | Trigger | Morphology | Example |
|---|---|---|---|
| Concentric hypertrophy | Pressure overload | ↑ wall thickness, normal/↓ cavity | Hypertension, aortic stenosis |
| Eccentric hypertrophy | Volume overload | ↑ cavity size, thin walls | Mitral/aortic regurgitation, DCM |
- Initially compensates by normalizing wall stress
- Eventually → diastolic dysfunction, ischemia, arrhythmias, systolic failure
4. Peripheral Vasoconstriction
- SNS + angiotensin II → arterial vasoconstriction → maintains blood pressure
- Also redistributes blood flow (away from skin/gut → toward brain/heart)
- Chronic: increases afterload → worsens cardiac output
5. Increased Heart Rate (Tachycardia)
- Sympathetic activation → sinus tachycardia compensates for reduced stroke volume: CO = HR × SV
- Chronic tachycardia → reduces diastolic filling time → worsens diastolic function
III. WHY COMPENSATION BECOMES MALADAPTIVE
The transition from compensated to decompensated heart failure occurs when:
- Neurohormonal activation exceeds benefit → increased myocardial oxygen demand, fibrosis, toxicity
- Ventricular remodeling reaches geometric limits
- Amplifying mechanisms perpetuate decompensation:
"Neurohormonal and inflammatory activation, ongoing myocardial injury with progressive myocardial dysfunction, worsening renal function, and interactions with the peripheral vasculature — all contribute to the propagation and worsening of the acute HF episode." — Braunwald's Heart Disease, p. 104
Summary Diagram (Conceptual)
Myocardial Injury / Overload
↓
↓ Cardiac Output / ↑ Filling Pressure
↓
┌───────────────────────────────────┐
│ COMPENSATION MECHANISMS │
│ • Frank-Starling (↑ preload) │
│ • SNS → tachycardia + inotropy │
│ • RAAS → Na+/H₂O retention │
│ • Ventricular hypertrophy │
└───────────────────────────────────┘
↓ (chronically)
Maladaptive Remodeling
Congestion + Low Output
→ Clinical Heart Failure
Sources: Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine, 12th Ed. (Elsevier); Textbook of Family Medicine, 9th Ed.
Typical manifestation of cardiovascular system dysfunction . Compensation mechanism..
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Excellent — rich content and diagrams retrieved. Now I'll compile the full, well-sourced answer.
Typical Manifestations of Cardiovascular System Dysfunction & Compensation Mechanisms
PART 1 — TYPICAL MANIFESTATIONS
Cardiovascular dysfunction ultimately results from two overlapping problems: reduced cardiac output (forward failure) and elevated filling pressures with congestion (backward failure).
"The primary signs and symptoms of all types of heart failure include tachycardia, decreased exercise tolerance, shortness of breath, and cardiomegaly. Peripheral and pulmonary edema are often but not always present. Decreased exercise tolerance with rapid muscular fatigue is the major direct consequence of diminished cardiac output. The other manifestations result from the attempts by the body to compensate for the intrinsic cardiac defect." — Katzung's Basic & Clinical Pharmacology, 16th Ed.
A. Left-Sided Heart Failure — Pulmonary Manifestations
The left ventricle fails to empty adequately → blood backs up into the pulmonary circulation → pulmonary venous hypertension and congestion.
| Manifestation | Mechanism |
|---|---|
| Dyspnea on exertion | ↑ pulmonary venous pressure → impaired gas exchange |
| Orthopnea | Supine position shifts fluid to lungs → ↑ pulmonary congestion |
| Paroxysmal nocturnal dyspnea (PND) | Same redistribution, worsened by nocturnal ↓ sympathetic tone |
| Pulmonary crackles (basal rales) | Fluid transudation into alveoli |
| Pleural effusion | Elevated pulmonary capillary pressure → fluid into pleural space |
| Pink frothy sputum | Severe pulmonary edema |
| Atrial fibrillation | Left atrial dilation from elevated LVEDP → atrial remodeling |
| Cough | Bronchial mucosal edema and irritation |
Morphology: The left ventricle is usually hypertrophied and often massively dilated. Secondary mitral incompetence and left atrial dilation are common, increasing the risk of atrial thrombus and AF. Microscopically: myocyte hypertrophy and interstitial fibrosis.
— Robbins, Cotran & Kumar: Pathologic Basis of Disease
B. Right-Sided Heart Failure — Systemic Congestion Manifestations
| Manifestation | Mechanism |
|---|---|
| Peripheral/dependent edema | ↑ systemic venous pressure → transcapillary fluid leak |
| Raised JVP (jugular venous pressure) | Backup of blood into systemic veins |
| Hepatomegaly / hepatic congestion | Elevated hepatic venous pressure |
| Ascites | Severe systemic venous hypertension |
| Anorexia / nausea | GI venous congestion and impaired gut motility |
| Nocturia | Redistribution of peripheral edema at night → ↑ renal perfusion |
C. General / Forward Failure Manifestations (Low Output)
| Manifestation | Mechanism |
|---|---|
| Fatigue & exercise intolerance | ↓ CO → skeletal muscle hypoperfusion |
| Tachycardia | Sympathetic compensation |
| Cardiomegaly | Ventricular hypertrophy and/or dilation |
| Hypotension / narrow pulse pressure | Severely ↓ stroke volume |
| Oliguria | Renal hypoperfusion → RAAS activation → avid Na⁺/H₂O retention |
| Cool, clammy extremities | Peripheral vasoconstriction from SNS/Ang II |
| Altered mental status | Cerebral hypoperfusion (severe/late) |
| Syncope / palpitations | Arrhythmias from ventricular remodeling |
| Cheyne-Stokes respiration | Periodic breathing from ↓ cerebral perfusion and ↑ CO₂ sensitivity |
PART 2 — COMPENSATION MECHANISMS
When cardiac output falls, the body activates a cascade of adaptive responses. These are initially beneficial but become maladaptive when sustained chronically.
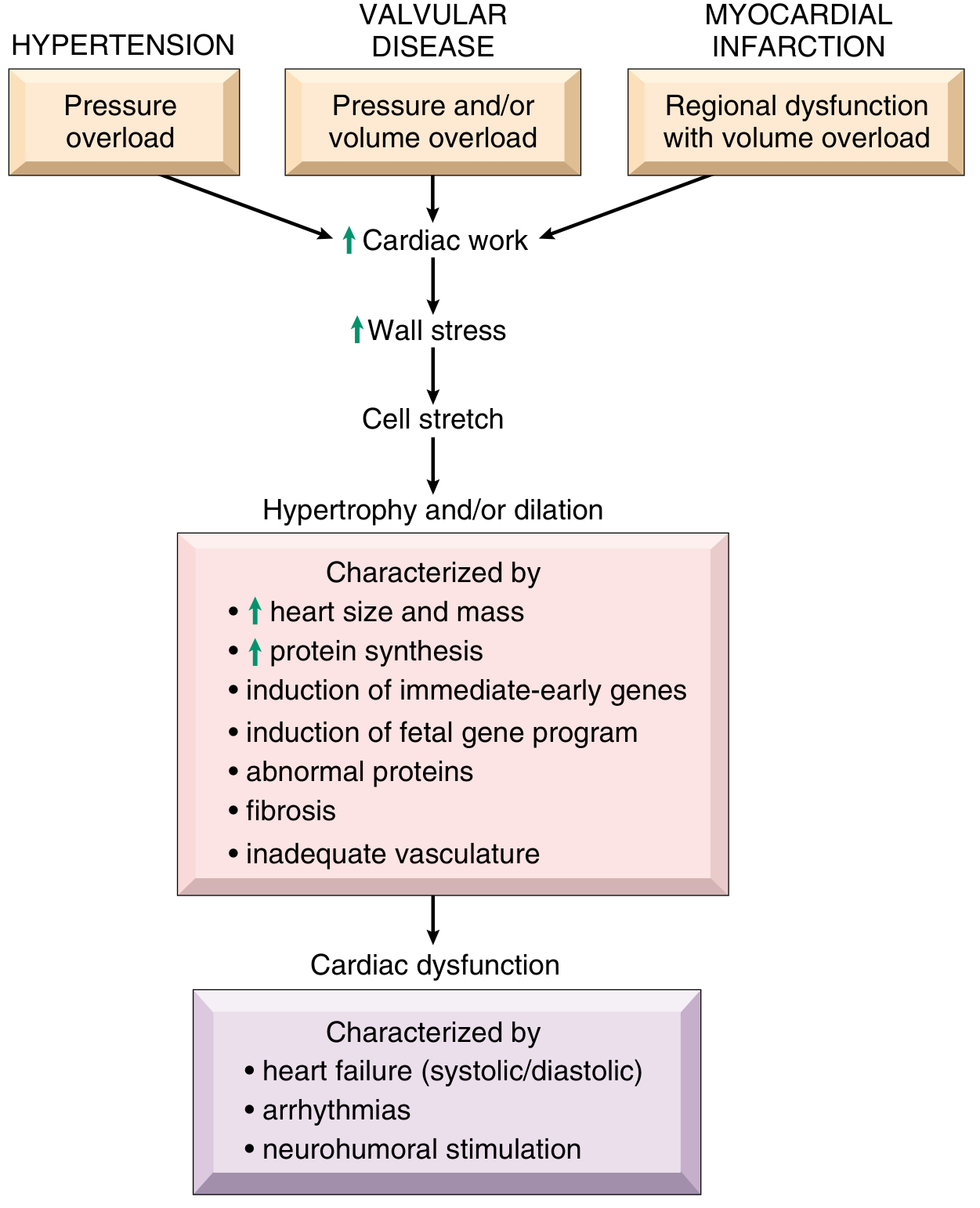
1. Frank-Starling Mechanism (Intrinsic)
- ↓ CO → incomplete ventricular emptying → ↑ end-diastolic volume (EDV) → greater myocyte stretch → ↑ force of contraction and stroke volume
- This is the ascending limb of the ventricular function curve
- At filling pressures >15 mmHg, a plateau is reached; beyond 20–25 mmHg → pulmonary congestion
- Benefit: immediate, no energy expenditure
- Limit: sarcomere over-stretch; the failing heart's function curve is shifted downward — the same filling pressure yields less output
2. Neurohumoral (Extrinsic) Compensation
This is the dominant systemic response. Two major arms:
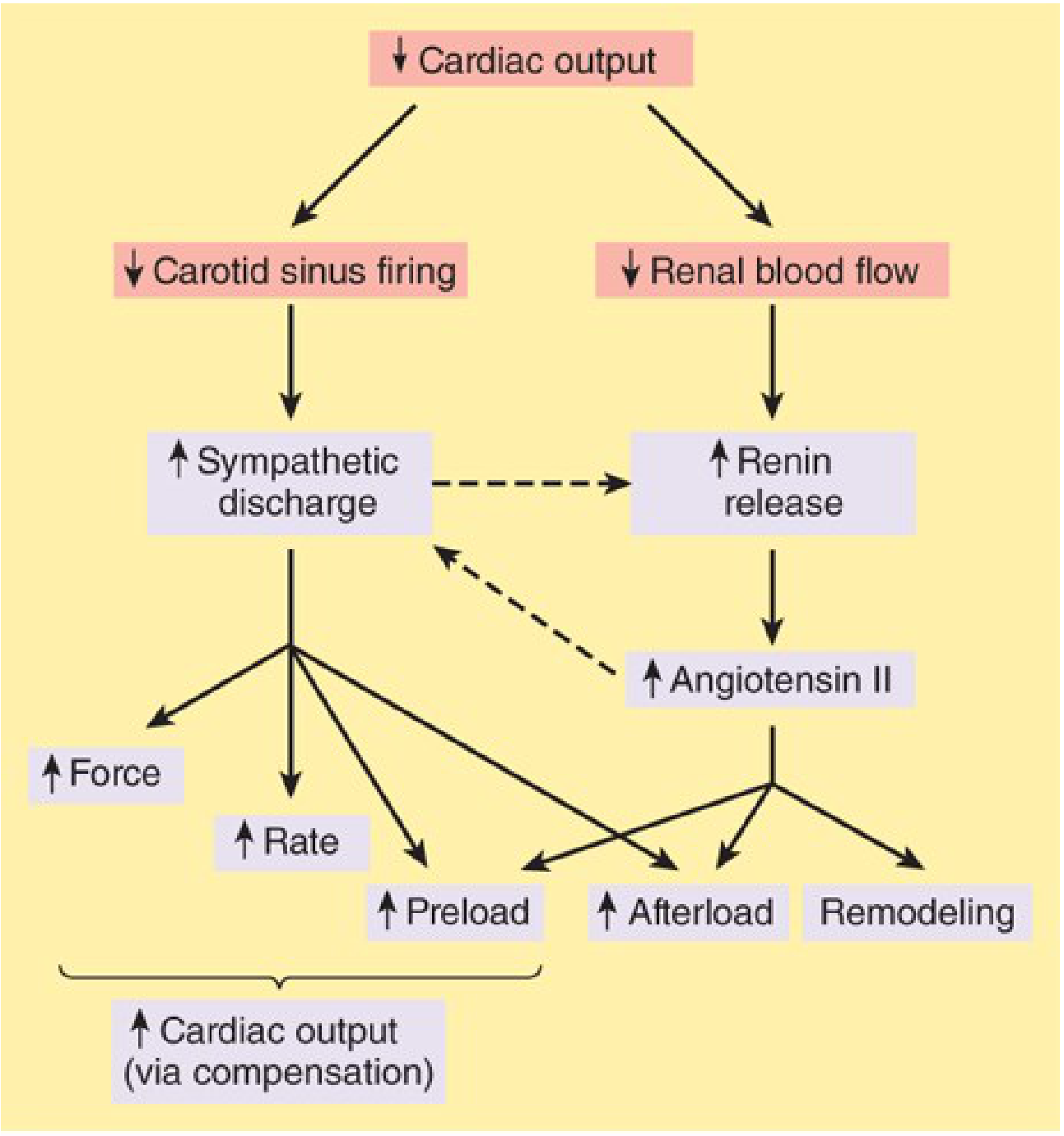
a. Sympathetic Nervous System (SNS)
- ↓ CO → ↓ carotid sinus baroreceptor firing → ↑ sympathetic outflow + ↓ parasympathetic outflow
- Effects: tachycardia, ↑ contractility (inotropy), arterial and venous vasoconstriction
- Short-term benefit: maintains perfusion pressure
- Chronic harm: β₁ receptor downregulation, myocyte apoptosis (via caspase activation), Ca²⁺ leak from sarcoplasmic reticulum via RyR channels → arrhythmias and ventricular stiffening
b. Renin-Angiotensin-Aldosterone System (RAAS)
- ↓ CO → ↓ renal perfusion → renin release → Angiotensin II formation
- Angiotensin II effects:
- Aldosterone secretion → Na⁺ and H₂O retention → ↑ preload
- Vasoconstriction → ↑ afterload
- Cardiac/vascular remodeling (fibrosis)
- Short-term benefit: volume expansion and pressure maintenance
- Chronic harm: ↑ afterload worsens EF; fibrosis impairs compliance; forms the vicious cycle of HF progression
c. Antidiuretic Hormone (ADH / Vasopressin)
- Released in response to ↑ Ang II and ↓ CO → free water retention → expands circulating volume
- Contributes to dilutional hyponatremia in advanced HF
d. Endothelin
- Potent vasoconstrictor released by vascular endothelium in response to ↓ perfusion → further increases afterload
e. Natriuretic Peptides (ANP, BNP) — Counter-regulatory
- Released by stretched atria (ANP) and ventricles (BNP) → vasodilation + natriuresis + diuresis
- Oppose RAAS and SNS activation
- NT-proBNP is used clinically as a diagnostic and prognostic biomarker for HF severity
"Sympathetic discharge facilitates renin release, and angiotensin II increases norepinephrine release by sympathetic nerve endings — forming a positive feedback that drives the vicious cycle of HF." — Katzung's Basic & Clinical Pharmacology, 16th Ed.
3. Ventricular Hypertrophy & Remodeling (Intrinsic)
"The most obvious intrinsic compensatory mechanism is myocardial hypertrophy. The increase in muscle mass helps maintain cardiac performance. However, after an initial beneficial effect, hypertrophy can lead to ischemic changes, impairment of diastolic filling, and alterations in ventricular geometry." — Katzung's Basic & Clinical Pharmacology, 16th Ed.
| Pattern | Stimulus | Sarcomere Assembly | Morphology | Example |
|---|---|---|---|---|
| Concentric hypertrophy | Pressure overload | Parallel (↑ cross-section) | ↑ wall thickness, normal/↓ cavity | Hypertension, aortic stenosis |
| Eccentric hypertrophy | Volume overload | In series (↑ length) | ↑ cavity, wall normal or thin | Mitral/aortic regurgitation, DCM |
Molecular changes in hypertrophy:
- Immediate-early genes (FOS, JUN, MYC) activated → ↑ protein synthesis
- Fetal gene program re-expressed (fetal myosin isoforms, natriuretic peptides, collagen)
- Fibrosis and inadequate capillary density → subendocardial ischemia
- Accelerated myocyte apoptosis → progressive dysfunction
Remodeling = slow structural changes including connective tissue proliferation and geometric distortion of the ventricle. Ultimately worsens systolic and diastolic function.
— Robbins, Cotran & Kumar
4. Peripheral Redistribution of Blood Flow
- SNS + Ang II → selective vasoconstriction → blood redistributed away from skin, gut, kidneys → towards brain and heart
- Short-term: preserves vital organ perfusion
- Chronic: renal underperfusion perpetuates RAAS; gut hypoperfusion → bacterial translocation and systemic inflammation
PART 3 — THE VICIOUS CYCLE (Why Compensation Fails)
↓ Cardiac Output
↓
SNS activation + RAAS activation
↓
↑ Heart rate ↑ Contractility ↑ Preload ↑ Afterload
↓ ↓
Short-term CO ↑ Ventricular remodeling
↓
Fibrosis, apoptosis, arrhythmias
↓
Progressive ↓ EF → Decompensated HF
The system reaches a new steady state where CO is lower and afterload is higher than optimal. This is the pathophysiological basis for why ACE inhibitors, ARBs, beta-blockers, aldosterone antagonists, and SGLT2 inhibitors — all neurohormonal antagonists — reduce mortality in heart failure.
Sources:
- Katzung's Basic & Clinical Pharmacology, 16th Edition — McGraw Hill
- Robbins, Cotran & Kumar: Pathologic Basis of Disease — Elsevier
- Goodman & Gilman's The Pharmacological Basis of Therapeutics — McGraw Hill
This is a shared conversation. Sign in to Orris to start your own chat.