Teach me everything I need to know as an MBBS student on chromosomal genetic disorders (Robbins pathology)
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Here is your complete MBBS-level teaching guide on chromosomal genetic disorders, synthesized directly from Robbins & Cotran Pathologic Basis of Disease and Robbins & Kumar Basic Pathology.
Chromosomal Genetic Disorders — Robbins Pathology
1. Background & Classification
Human genetic diseases fall into three broad categories:
- Monogenic (Mendelian) disorders — single gene mutations with large effect, follow classic inheritance
- Chromosomal disorders — abnormal number or structure of chromosomes (this chapter)
- Complex multigenic disorders — multiple genes + environment (diabetes, hypertension)
Key general principles (memorize these):
- Loss of chromosomal material → more severe defects than gain
- Autosomal monosomy → generally lethal (too much gene loss); autosomal trisomy → less severe but still major
- Sex chromosome imbalances are tolerated much better than autosomal ones — explained by lyonization and the Y chromosome carrying little genetic information
- Sex chromosome disorders are often subtle, not detected at birth; infertility is the commonest presentation
- Most chromosomal disorders arise de novo (parents are normal; recurrence risk in siblings is low) — except translocation Down syndrome
2. The Normal Karyotype & Cytogenetic Basics
2.1 How Karyotyping is Done
- Dividing cells arrested in metaphase using mitotic spindle inhibitors (e.g., colchicine)
- Stained with Giemsa (G-banding) → alternating light and dark bands
- Standard G-banding resolves ~400–800 bands per haploid set; prophase banding → up to 1500 bands
- Normal: 46,XX (female), 46,XY (male)
Fig. 5.19 — Normal male G-banded karyotype (46,XY)
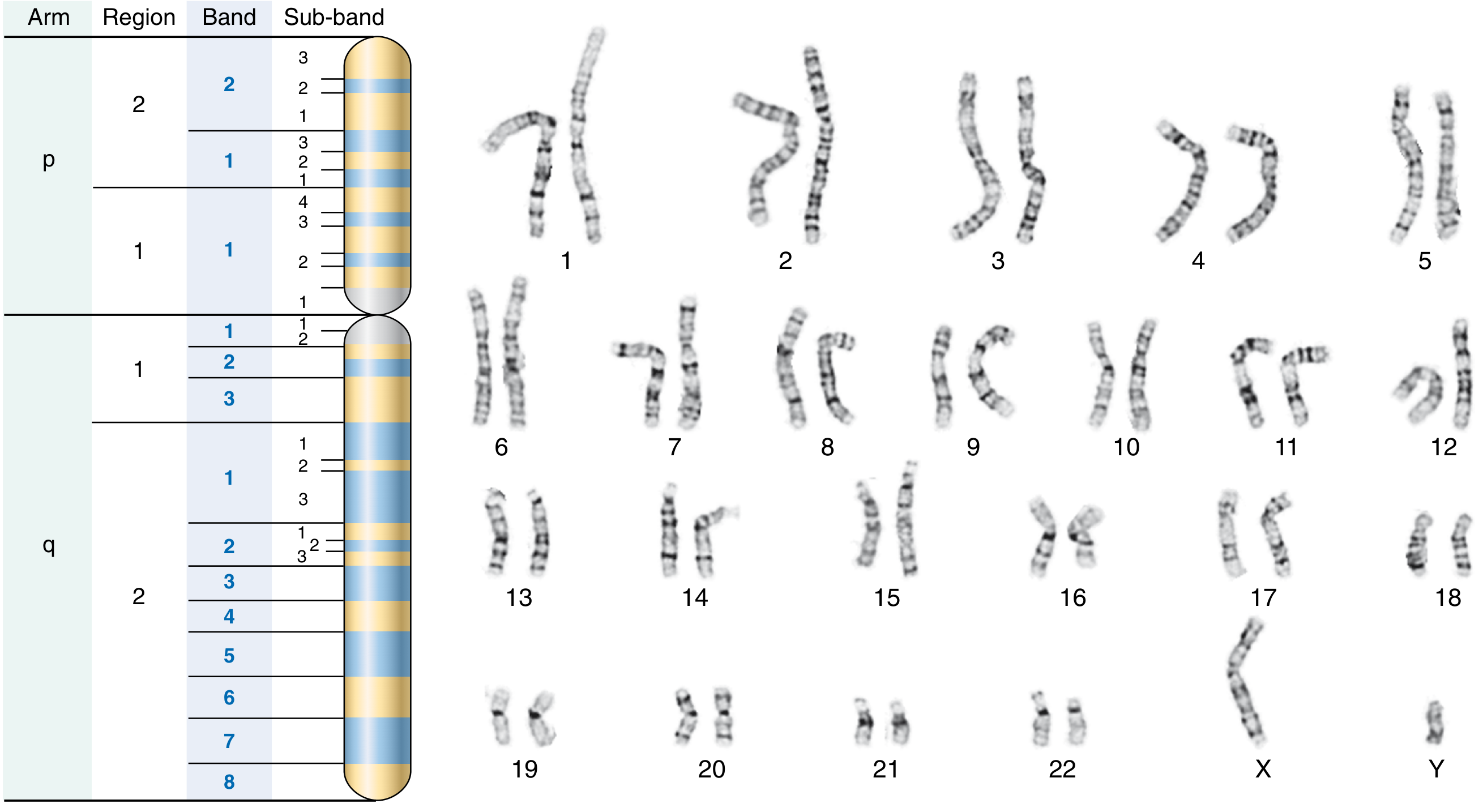
2.2 Cytogenetic Notation
| Notation | Meaning |
|---|---|
| p | Short arm (from French petit) |
| q | Long arm |
| + or − before chromosome number | Extra or missing whole chromosome |
| t(A;B) | Translocation between chromosomes A and B |
| del | Deletion |
| i | Isochromosome |
| r | Ring chromosome |
Example: A male with trisomy 21 → 47,XY,+21
A 22q11.2 deletion: region on long arm of chr 22, region 1, band 1, sub-band 2
3. Types of Chromosomal Abnormalities
3.1 Numerical Abnormalities
| Term | Definition |
|---|---|
| Euploid | Exact multiple of haploid number (23): normal (46), triploidy (69), tetraploidy (92) |
| Aneuploid | NOT an exact multiple of 23 |
| Trisomy | 2n + 1 (47 chromosomes) |
| Monosomy | 2n − 1 (45 chromosomes) |
Mechanisms:
- Nondisjunction — failure of chromosomes to separate during meiosis I or II (or mitosis) → gametes with n+1 or n−1 chromosomes
- Anaphase lag — a chromosome lags behind and is excluded from the nucleus → one normal cell + one monosomic cell
Mosaicism — two or more genetically distinct cell populations in one individual, from mitotic nondisjunction early in embryogenesis. Phenotype is milder, proportional to the abnormal cell fraction.
3.2 Structural Abnormalities
| Type | Description | Notation Example |
|---|---|---|
| Balanced reciprocal translocation | Single break in two chromosomes, exchange of segments; carrier has 46 chromosomes but altered morphology; phenotypically normal but produces aneuploid offspring | 46,XX,t(2;5)(q31;p14) |
| Robertsonian translocation | Fusion of long arms of two acrocentric chromosomes (13, 14, 15, 21, 22); the short arms are lost; carrier has only 45 chromosomes | 45,XX,der(14;21)(q10;q10) — important in familial Down syndrome |
| Deletion | Loss of a chromosomal segment; can be interstitial or terminal | del(22)(q11.2) |
| Inversion | Segment removed, flipped 180°, reinserted; paracentric (doesn't include centromere) or pericentric (includes centromere) | |
| Ring chromosome | Breaks at both ends, ends join; unstable | r(X) |
| Isochromosome | Faulty division at centromere → two identical arms; most common: i(Xq) — two long arms of X | i(X)(q10) |
Pearl: Balanced structural rearrangements → phenotypically normal carriers, but produce aneuploid gametes at meiosis (risk of affected offspring). Unbalanced rearrangements → phenotypic abnormalities.
4. Cytogenetic Disorders Involving Autosomes
4.1 Trisomy 21 — Down Syndrome ⭐ (Most Important)
Karyotypes:
| Type | Frequency | Key Point |
|---|---|---|
| Free trisomy 21 | ~95% | Meiotic nondisjunction; maternal age effect; parents normal karyotype |
| Robertsonian translocation | ~4% | Translocation of chr 21q onto chr 14 or 22; carrier parent (usually mother, 45 chromosomes); familial in some; no maternal age effect |
| Mosaicism | ~1% | Mitotic nondisjunction post-fertilization; milder phenotype; no maternal age effect |
Maternal age effect:
- < 20 years: 1 in 1550 live births
-
45 years: 1 in 25 live births
- In 95% of cases, the extra chr 21 is of maternal origin
FISH diagnosis of trisomy 21:
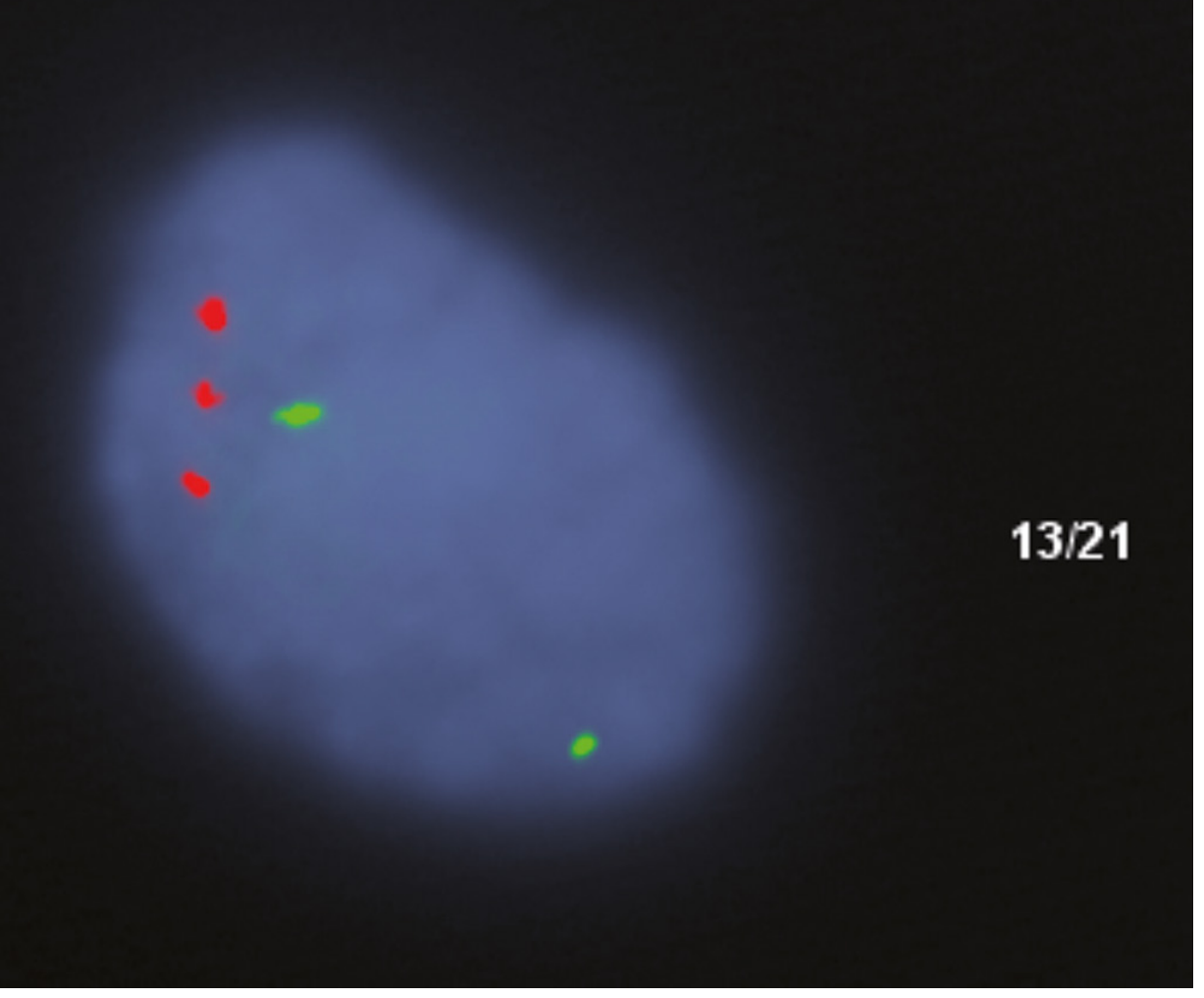 Three red signals = trisomy 21; two green signals = normal chr 13
Three red signals = trisomy 21; two green signals = normal chr 13
Clinical features — Down Syndrome:
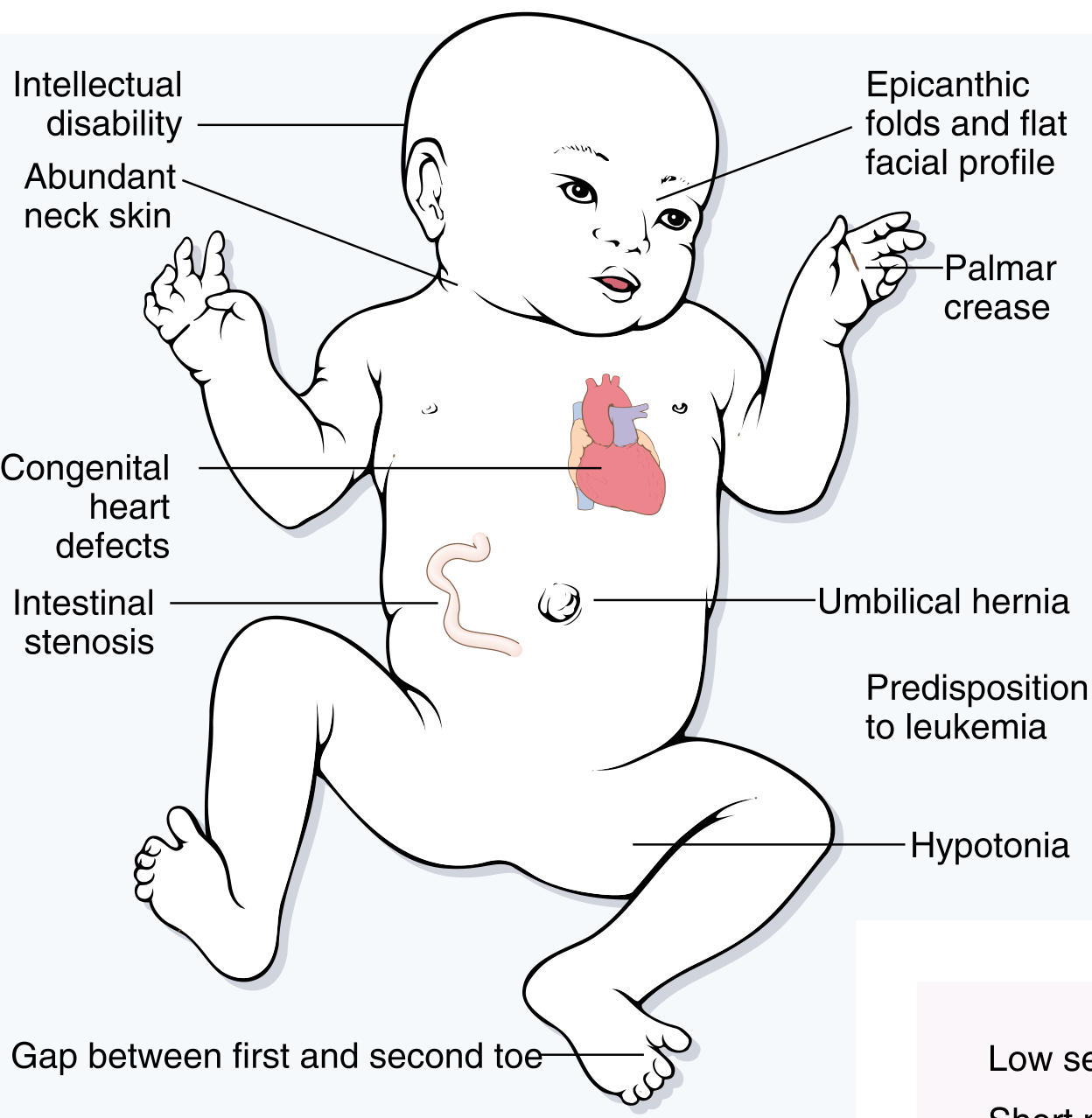
| System | Features |
|---|---|
| Facies | Flat facial profile, oblique palpebral fissures, epicanthic folds, Brushfield spots (iris), protruding tongue, small ears |
| Neurologic | Intellectual disability (IQ 25–50 in most); hypotonia |
| Cardiac | ~40% have congenital heart disease — most common: AV septal defect (43%), then VSD (32%), ASD (19%), tetralogy of Fallot; cardiac disease = leading cause of death in infancy |
| GI | Intestinal stenosis/atresia (duodenal atresia), umbilical hernia, Hirschsprung disease |
| Hands | Single palmar crease (simian crease), wide gap between 1st and 2nd toes, short broad hands |
| Hematologic | 10–20× increased risk of leukemia (ALL in children; AML — notably transient myeloproliferative disorder in neonates) |
| Endocrine | Hypothyroidism (autoimmune) |
| Neurodegeneration | Virtually all patients >40 years develop Alzheimer disease (APP gene on chr 21 encodes amyloid precursor protein) |
| Immunity | Increased infections (impaired T and B cell function) |
| Fertility | Males: infertile; Females: may be fertile |
Life expectancy: ~60 years (cardiac disease is the main killer in early life)
Prenatal diagnosis:
- Cell-free fetal DNA (cfDNA) from maternal blood → next-generation sequencing → highly sensitive noninvasive screen
- Confirmed by conventional karyotyping of chorionic villus sample or amniotic fluid
Pathogenesis: Overexpression of genes on chr 21 (dosage effect) including:
- APP → Alzheimer disease
- SOD1 → excess free radicals (mitochondrial dysfunction)
- Highest density of lncRNAs of any chromosome (functions largely unknown)
4.2 Trisomy 18 — Edwards Syndrome
| Feature | Detail |
|---|---|
| Karyotype | 47,XX or 47,XY,+18 |
| Incidence | 1 in 8000 births |
| Cause | Meiotic nondisjunction; maternal age effect |
| Severity | Severe, wide-ranging malformations |
| Survival | Rarely > 1 year; most die within weeks to months |
Clinical features:
- Rocker-bottom feet, overlapping fingers (index finger over 3rd, 5th over 4th)
- Micrognathia, prominent occiput
- Severe congenital heart defects
- Intellectual disability, hypertonia
- Renal malformations
4.3 Trisomy 13 — Patau Syndrome
| Feature | Detail |
|---|---|
| Karyotype | 47,XX or 47,XY,+13 |
| Incidence | Rarer than trisomy 18 |
| Severity | Most severe of the three common trisomies |
| Survival | Most die within weeks to months |
Clinical features (the "midline" defects):
- Holoprosencephaly (failure of forebrain division)
- Cleft lip/palate
- Microphthalmia/anophthalmia, cyclopia
- Polydactyly
- Congenital heart defects (VSD, ASD, PDA)
- Cutis aplasia (absent skin patches on scalp — pathognomonic)
4.4 Chromosome 22q11.2 Deletion Syndrome (DiGeorge / Velocardiofacial)
| Feature | Detail |
|---|---|
| Karyotype | del(22)(q11.2) — small deletion of ~1.5 Mb, 30–40 genes |
| Incidence | ~1 in 4000 births; often missed due to variable presentation |
| Diagnosis | FISH (90% sensitivity for DiGeorge, 80% for velocardiofacial) |
Key gene: TBX1 (T-box transcription factor) — expressed in pharyngeal mesenchyme/endodermal pouch → controls development of face, thymus, parathyroid. Targets include PAX9.
Two clinical faces of the same deletion:
| DiGeorge Syndrome | Velocardiofacial Syndrome |
|---|---|
| Thymic hypoplasia → T-cell immunodeficiency | Facial dysmorphism (prominent nose, retrognathia) |
| Hypocalcemia (parathyroid hypoplasia) | Cleft palate |
| Cardiac outflow tract defects | Cardiovascular anomalies |
| Mild facial anomalies | Learning disabilities |
Additional features common to both:
- Schizophrenia in ~25% of adults (2–3% of childhood-onset schizophrenia have this deletion)
- ADHD in 30–35% of affected children
- Atopic disorders, autoimmunity
Pearl: 30% of individuals with conotruncal cardiac defects alone also harbor 22q11.2 deletions.
5. Cytogenetic Disorders Involving Sex Chromosomes
Why sex chromosome disorders are better tolerated — Lyon Hypothesis
Mary Lyon (1962): In females, only one X chromosome is genetically active per cell. X inactivation:
- Occurs early in fetal life (~16 days post-conception)
- Random — either maternal or paternal X is silenced in each cell
- All progeny of that cell maintain the same inactive X (clonal)
- Molecular basis: XIST gene → encodes a long noncoding RNA that "coats" the X chromosome from which it is transcribed → silences genes on that X; the other XIST allele is switched off in the active X
Important caveats:
- Not all genes on the inactive X are silent: ~30% of genes on Xp and ~3% on Xq escape X inactivation — this explains phenotypic effects in Turner syndrome (monosomy X)
- All X chromosomes beyond one are inactivated, so even 48,XXXX females have only one active X
- Y chromosome carries little genetic information — the critical gene is SRY (sex-determining region Y) on Yp → dictates male phenotype regardless of number of X chromosomes present
5.1 Klinefelter Syndrome ⭐
| Feature | Detail |
|---|---|
| Definition | Male hypogonadism with ≥2 X chromosomes + ≥1 Y chromosome |
| Karyotype | Most common: 47,XXY; mosaics: 46,XY/47,XXY (milder); more severe: 48,XXXY, 49,XXXXY |
| Cause | Meiotic nondisjunction; maternal and paternal nondisjunction contribute equally |
| Incidence | One of the most common causes of hypogonadism in males (~1 in 500–1000 male births) |
Clinical features:
| Feature | Detail |
|---|---|
| Body habitus | Tall, elongated lower limbs (floor-to-pubis > pubis-to-crown) |
| Testes | Small, firm (as little as 2 cm); atrophic tubules with hyalinization |
| Gonadotropins | Elevated FSH; reduced testosterone |
| Gynecomastia | Present |
| Hair | Reduced facial, body, and pubic hair |
| Fertility | Virtually always sterile due to azoospermia (impaired spermatogenesis); rarely fertile mosaics |
| Intellect | Usually normal, but verbal IQ may be slightly reduced; learning difficulties in some |
| Risk | Increased risk of breast cancer (comparable to females), extragonadal germ cell tumors, autoimmune diseases |
Pathology of testes: Hyalinization of seminiferous tubules → only Sertoli cells remain → no spermatozoa. Leydig cells initially appear prominent (pseudo-hyperplasia).
Why multiple X chromosomes cause hypogonadism: The extra X chromosomes, though largely inactivated, still express pseudoautosomal and other genes that escape X inactivation → interfere with normal testicular development.
5.2 Turner Syndrome ⭐
| Feature | Detail |
|---|---|
| Definition | Female hypogonadism from complete or partial monosomy X |
| Incidence | Most common sex chromosome disorder in females; single most important cause of primary amenorrhea (~1/3 of all cases) |
Karyotypes (highly variable):
| Type | Frequency | Karyotype |
|---|---|---|
| Monosomy X | ~57% | 45,X |
| Structural X abnormalities | ~14% | Isochromosome of Xq: 46,X,i(X)(q10); ring: 46,X,r(X); deletions: 46,X,del(Xq) or 46,X,del(Xp) |
| Mosaics | ~29% (likely higher with sensitive techniques — up to 75%) | 45,X/46,XX; 45,X/46,XY; 45,X/47,XXX; 45,X/46,X,i(X)(q10) |
Pearl: Mosaics with a high proportion of 46,XX cells may have nearly normal appearance and present only with primary amenorrhea. A very small number can conceive.
Pearl: 5–10% of Turner patients have Y chromosome sequences (45,X/46,XY or Y fragments) → significantly higher risk of gonadoblastoma → prophylactic gonadectomy indicated.
Why monosomy X causes phenotypic effects despite X inactivation: Because ~30% of genes on Xp escape X inactivation → haploinsufficiency for these genes → phenotype. Key gene: SHOX (short stature homeobox gene) on Xp → explains short stature.
Clinical features:
In infancy/neonates:
- Lymphedema of dorsum of hands and feet
- Cystic hygroma (distended lymphatics at nape of neck) → may be detected on prenatal US
- Webbed neck (pterygium colli) — residual from resolved hygroma
Cardiovascular — most important cause of mortality in children:
- Coarctation of aorta (preductal) — ~5% of females with coarctation have Turner syndrome
- Bicuspid aortic valve (most common cardiac anomaly overall)
- Aortic root dilation (30%); aortic dissection risk ×100 vs. normal
At puberty and in adults:
- Short stature (rarely >150 cm) — most consistent feature
- Primary amenorrhea and failure to develop secondary sex characteristics
- Streak gonads (bilateral fibrous streaks replacing ovaries) → infertility
- Infantile genitalia, poor breast development, scant pubic hair
- Shield chest, widely spaced nipples
- Low posterior hairline, high arched palate
- Cubitus valgus (wide carrying angle)
- Normal intellect but subtle deficits in nonverbal/visual-spatial processing
- ~50% develop autoimmune thyroiditis (hypothyroidism)
- Glucose intolerance, obesity
5.3 Other Sex Chromosome Aneuploidies (Brief)
| Karyotype | Phenotype |
|---|---|
| 47,XXX (Triple X) | Phenotypically normal female; may have mild learning difficulties; fertile in most cases; may have menstrual irregularities |
| 47,XYY | Tall males; normal fertility (usually); mild behavioral/learning issues; NOT associated with increased criminal behavior (this old teaching is incorrect) |
| 48,XXXY / 49,XXXXY | Progressively more severe Klinefelter-like features; intellectual disability increases with extra X chromosomes |
6. Hermaphroditism and Pseudohermaphroditism (Brief Overview)
| Term | Definition |
|---|---|
| True hermaphrodite | Both ovarian and testicular tissue present (either in one gonad = ovotestis, or separately) |
| Female pseudohermaphrodite (46,XX) | Genetic female; virilized external genitalia; e.g., congenital adrenal hyperplasia (21-hydroxylase deficiency — not chromosomal) |
| Male pseudohermaphrodite (46,XY) | Genetic male; incompletely masculinized; e.g., androgen insensitivity syndrome (testicular feminization) |
The key point for chromosomal disorders: the SRY gene on Yp is the master switch for male differentiation. Its absence → female phenotype regardless of other chromosomes. Rare XX males exist from SRY translocation to an X chromosome.
7. Summary Comparison Table — High-Yield for Exams
| Disorder | Karyotype | Key Mechanism | Hallmark Features | Key Complication |
|---|---|---|---|---|
| Down Syndrome | 47,XY/XX,+21 | Meiotic nondisjunction (95%); Robertsonian translocation (4%); Mosaicism (1%) | Flat facies, epicanthic folds, intellectual disability, hypotonia, simian crease | AV septal defect (40%); Alzheimer disease (>40 yrs); Leukemia |
| Edwards Syndrome | 47,+18 | Meiotic nondisjunction | Rocker-bottom feet, overlapping fingers, micrognathia | Death < 1 year |
| Patau Syndrome | 47,+13 | Meiotic nondisjunction | Holoprosencephaly, cleft lip/palate, polydactyly, cutis aplasia | Death < weeks–months |
| 22q11.2 deletion | del(22q11.2) | Microdeletion; TBX1 haploinsufficiency | Thymic aplasia (T-cell deficiency), hypocalcemia, conotruncal cardiac defects | Schizophrenia (25%) |
| Klinefelter | 47,XXY | Meiotic nondisjunction (equal maternal/paternal) | Tall, small firm testes, gynecomastia, azoospermia | Sterility; breast cancer risk |
| Turner Syndrome | 45,X (57%); Mosaic (29%); Structural X (14%) | Monosomy X (various mechanisms) | Short stature, webbed neck, primary amenorrhea, streak gonads, coarctation of aorta | Aortic dissection; gonadoblastoma (if Y material present) |
8. Diagnostic Methods — Cytogenetic & Molecular
| Method | What it Detects | Resolution |
|---|---|---|
| Conventional karyotyping (G-banding) | Numerical + large structural aberrations | ~400–800 bands; ~4–10 Mb |
| FISH | Specific deletions, duplications, translocations; can be done on interphase cells | Down to kilobases |
| Array-CGH (chromosomal microarray) | Copy number variants genome-wide | ~50–100 kb |
| Next-generation sequencing (NGS) | Single nucleotide variants, small indels, copy number variants | Single nucleotide |
| Prenatal cfDNA (NIPT) | Fetal aneuploidy from maternal blood | Screening (confirm with karyotyping) |
| PCR | Specific sequence alterations, trinucleotide repeats | DNA/RNA level |
9. Key Exam Pearls (Robbins)
- Nondisjunction is the most common cause of aneuploidy; anaphase lag produces monosomy only
- Monosomy of an autosome is almost always lethal; trisomies 21, 18, 13 survive
- Maternal age matters for free trisomies (nondisjunction); it does NOT matter for translocation or mosaic Down syndrome
- Robertsonian translocation: carrier has 45 chromosomes but normal phenotype; risk of Down syndrome offspring is high
- Turner syndrome = most common cause of primary amenorrhea; 45,X is the most frequent karyotype
- Klinefelter = most common cause of male hypogonadism/infertility due to chromosomal cause
- Lyon hypothesis: X-inactivation starts day 16; mediated by XIST lncRNA; ~30% of Xp genes escape inactivation
- 22q11.2 deletion: diagnose with FISH; TBX1 is the key gene; psychiatric risk = 25% schizophrenia
- All Down syndrome patients >40 years → Alzheimer disease (APP gene on chr 21)
- Y-material in Turner syndrome → gonadoblastoma risk → gonadectomy
Sources: Robbins, Cotran & Kumar — Pathologic Basis of Disease, Chapter 5 (pp. 144–175); Robbins & Kumar Basic Pathology, Chapter 4 (pp. 144–165)
Teach me everything I need to know as an MBBS student on Single Gene Genetic Disorders (Robbins Pathology)
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.