Acute lymphoid leukaemia
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute lymphoblastic leukemia"[MeSH Terms] AND treatment
Finding Sources
Finding Sources
Searching PubMed
"acute lymphocytic leukemia" AND treatment
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, authoritative content from multiple major textbooks. Let me compile the complete answer.
Acute Lymphoid (Lymphoblastic) Leukaemia (ALL)
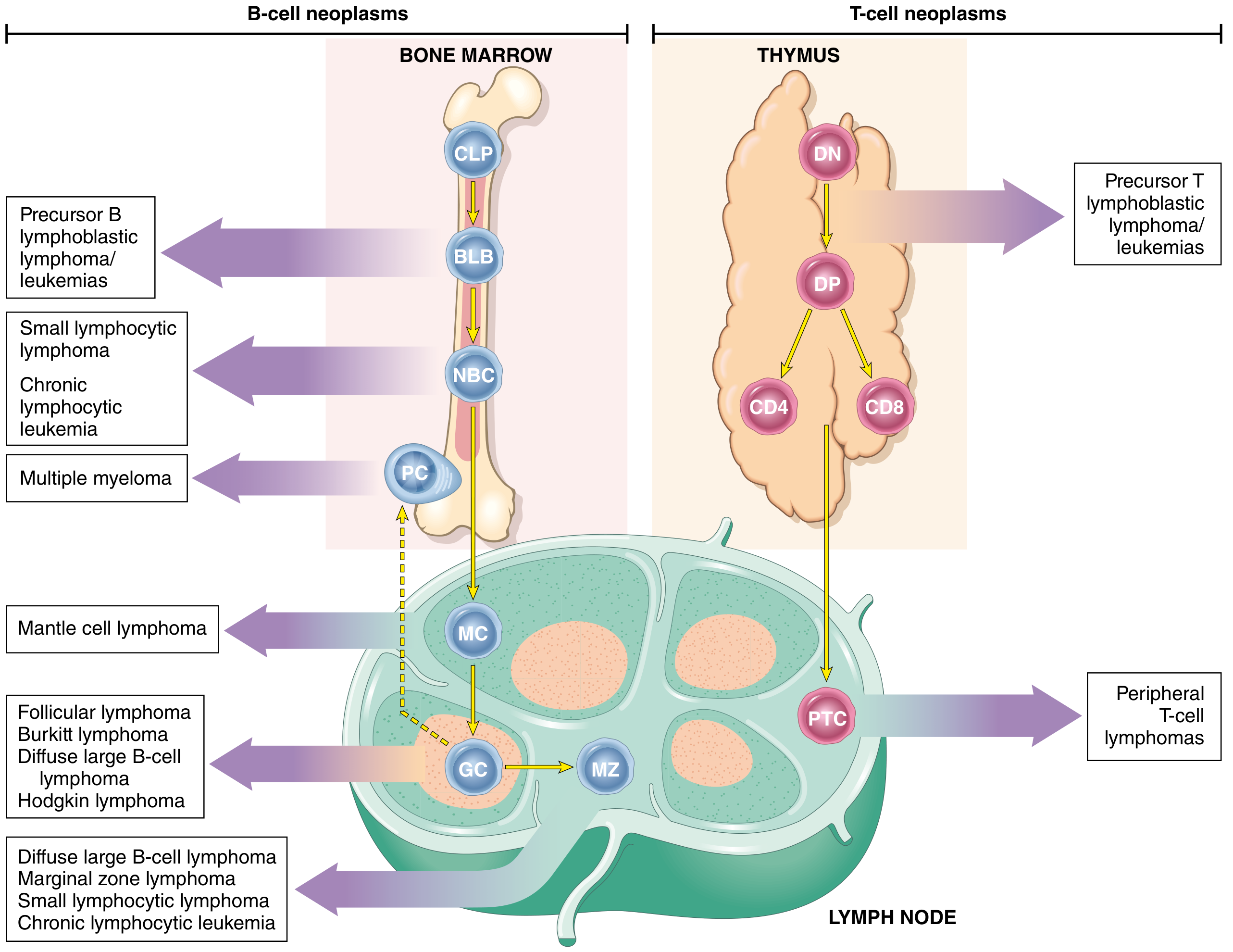
Definition
ALL is a neoplasm of immature B (pre-B) or T (pre-T) lymphocytes called lymphoblasts, arrested at an early stage of differentiation in the bone marrow or lymphatic system, resulting in the accumulation of non-functional leukemic cells that displace normal haematopoiesis.
- ~85% are B-ALLs
- ~15-25% are T-ALLs
Epidemiology
- Most common cancer in children, with a peak incidence at 3-4 years of age
- B-ALL peaks at ~3 years (paralleling the maximum number of normal bone marrow pre-B cells); T-ALL peaks in adolescence (when the thymus reaches maximum size)
- Slightly more frequent in boys than girls
- Hispanic/Latino children have the highest incidence of any ethnic group in the US
- Adult incidence: 0.7-1.8/100,000/year; rises again in the elderly
- Ph+ ALL accounts for ~50% of elderly B-lineage ALL patients
Etiology & Risk Factors
| Factor | Detail |
|---|---|
| Ionizing radiation | Increased risk, more so for AML |
| Prior chemotherapy | Alkylating agents, topoisomerase inhibitors |
| Congenital disorders | Down syndrome (20-fold increased risk), Klinefelter syndrome, Fanconi anaemia, Bloom syndrome, ataxia-telangiectasia, neurofibromatosis |
| Viruses | EBV (Burkitt type); HTLV-I (adult T-cell leukaemia/lymphoma) |
Pathogenesis & Molecular Biology
Most ALLs carry chromosomal aberrations that dysregulate transcription factors needed for normal lymphocyte development. Fewer than 10 driver mutations are generally sufficient.
Key genetic alterations in B-ALL:
| Mutation | Frequency | Significance |
|---|---|---|
| t(9;22) Philadelphia chromosome (BCR-ABL1) | ~5% children, ~25% adults, ~50% elderly | Constitutive ABL1 kinase activation; TKI-targetable |
| t(12;21) ETV6-RUNX1 | ~25% children | Favourable prognosis |
| Hyperdiploidy (>50 chromosomes) | B-ALL only | Better prognosis |
| Hypodiploidy | B-ALL only | Worse prognosis |
| t(4;11) KMT2A-AF4 | ~7% adults | Poor prognosis |
| t(8;14) MYC-IGH | Burkitt variant | Mature B-cell ALL |
| PAX5 mutations | ~30% B-ALL | B-cell maturation arrest |
| IKZF1 mutations | ~25% B-ALL | Poor prognosis |
| CRLF2 + JAK2 mutations | Ph-like ALL (20-25% adults) | Poor prognosis, TKI-targetable |
Key genetic alterations in T-ALL:
- NOTCH1 mutations: 50-70% of T-ALL (essential for T-cell development)
- Translocations at T-cell receptor enhancer regions (chromosomes 7 and 14)
"Ph-like ALL" lacks classic BCR-ABL1 but has kinase-activating mutations in the same signalling pathway - accounting for 20-25% of adult ALLs and carrying a poor prognosis.
Classification
Immunophenotype (B-cell lineage, ~75% of ALL)
| Subtype | Key Markers | Frequency |
|---|---|---|
| Pro-B (null) ALL | HLA-DR, TdT, CD19; no CD10 | ~10-12% adults |
| Common ALL (CALLA+) | CD19, CD22, CD10 (CALLA) | ~50-60% of ALL |
| Pre-B ALL | CD10 + cytoplasmic Ig | ~10% |
| Mature B-ALL (Burkitt) | Surface IgM | ~3-5% adults |
Best therapeutic outcomes among B-cell types: CALLA-positive ALL
T-cell lineage (~25% of ALL)
| Subtype | Key Markers |
|---|---|
| Early T-cell precursor (ETP-ALL) | CD7+, CD1a-, surface CD3- |
| Thymic T-ALL | CD1a+ |
| Mature T-ALL | Surface CD3+ |
The WHO now classifies ALL primarily on cytogenetic and molecular features rather than morphology alone.
FAB Morphology (historical)
- L1: Small blasts (common in children)
- L2: Large heterogeneous blasts (common in adults)
- L3: Vacuolated blasts = Burkitt leukaemia (mature B-ALL); ~5% of adults
Clinical Features
Signs and symptoms result from marrow failure (replacement of normal haematopoiesis) and organ infiltration:
Marrow failure:
- Anaemia - fatigue, pallor, headache, dyspnoea
- Thrombocytopenia - petechiae, ecchymoses, bleeding gums, epistaxis (~1/3 of patients have clinically evident bleeding at diagnosis)
- Granulocytopenia - bacterial infections (~1/3 have significant or life-threatening infections at presentation)
Organ infiltration (more pronounced in ALL than AML):
- Lymphadenopathy, hepatosplenomegaly - common at diagnosis
- Mediastinal mass - characteristic of T-ALL (thymic origin)
- Bone pain - from periosteal infiltration or marrow expansion; especially in children
- CNS involvement (leukemic meningitis) - headache, nausea, cranial nerve palsies
- Leukemia cutis - raised, non-pruritic rash
- Testicular infiltration (especially in relapsed childhood ALL)
Diagnosis
Peripheral Blood
Initial workup includes CBC with differential (Wright-Giemsa stain).
| Lab Parameter | Common finding at diagnosis |
|---|---|
| WBC < 10 × 10⁹/L | 41% |
| WBC 10-50 × 10⁹/L | 31% |
| WBC > 50 × 10⁹/L | 28% |
| WBC > 100 × 10⁹/L | 16% |
| Neutropenia, anaemia, thrombocytopenia | Nearly universal |
Bone Marrow
-
20% blasts required for diagnosis
- Morphology, cytochemistry (TdT+, Sudan black-, MPO- in ALL)
Immunophenotyping (flow cytometry) - essential
- B-lineage: CD19, CD22, CD10, CD20, cytoplasmic/surface Ig
- T-lineage: CD7, CD1a, CD3 (surface/cytoplasmic)
- A marker considered positive if >20% cells stain positive
Cytogenetics / Molecular
- Karyotype, FISH, PCR for BCR-ABL1, KMT2A rearrangements, etc.
- Identifies prognostic subgroups and therapy targets
CSF Examination (Lumbar Puncture)
- Essential in all ALL patients
- CNS involvement defined as ≥5 cells/μL or leukemic blasts on morphology
- Intrathecal methotrexate should be given at the first LP to eliminate any transferred blast cells
- Only in patients with adequate platelets (>20 × 10³/L) and without active haemorrhage
Antigen Targets for Immunotherapy in B-ALL
| Antigen | Expression | Monoclonal Antibody |
|---|---|---|
| CD20 | 86-100% (Burkitt); 30-40% (B-precursor) | Rituximab, Ofatumumab |
| CD22 | 93-98% (B-precursor); ~100% (mature B) | Inotuzumab ozogamicin, Epratuzumab |
| CD19 | 95-100% (B-precursor and mature) | Blinatumomab; CAR-T cells |
(Harrison's Principles of Internal Medicine, 22nd Ed.)
Treatment
Treatment is divided into three phases:
1. Induction (3-4 weeks)
Goal: complete remission (blasts undetectable, normal marrow restored)
Core regimen (all adults): vincristine + prednisone/dexamethasone + L-asparaginase + daunorubicin
- CR achieved in 90% of children and 80-90% of adults
- Dexamethasone preferred over prednisone (better CNS penetration, acts on resting blasts)
- CD20-positive, Ph-negative ALL: add rituximab (375 mg/m²) - improves outcome
- Two widespread adult regimens:
- BFM (Berlin-Frankfurt-Münster) protocol - common in Europe
- Hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) - common in North America
2. Post-remission Consolidation (6-8 courses)
Without further therapy, virtually 100% relapse.
- High-dose methotrexate (1-5 g/m²) and/or high-dose cytarabine (1-3 g/m²) - essential for sanctuary site penetration (CNS)
- Ph+ ALL: add a TKI (imatinib, dasatinib, ponatinib)
- Allogeneic stem cell transplantation (SCT) for high-risk patients in first CR
3. Maintenance Therapy (2-2.5 years)
- 6-mercaptopurine (daily) + methotrexate (weekly) + intrathecal therapy
- For Ph+ ALL: add a TKI throughout maintenance; continue 2-2.5 years guided by MRD
- Exception: Burkitt leukaemia requires only 6-12 months maintenance
CNS Prophylaxis and Treatment
- Intrathecal methotrexate is standard
- High-dose systemic methotrexate/cytarabine reaches CNS sanctuary sites
- Cranial irradiation now largely replaced by intrathecal chemotherapy to avoid neurocognitive side effects
Ph+ ALL - Special Considerations
- BCR-ABL1 fusion (190 kD in ALL vs. 210 kD in CML)
- TKI (imatinib, dasatinib, ponatinib) added to chemotherapy - dramatically improves outcomes
- A chemotherapy-free induction with dasatinib + dexamethasone + blinatumomab achieves:
- CR rate 98%
- 2-year OS 95%, DFS 88%
- Blinatumomab eliminates Ph+ cells with TKI-resistant mutations
Immunotherapy & Novel Agents
| Agent | Class | Target | Key Use |
|---|---|---|---|
| Blinatumomab | Bispecific antibody (CD19 × CD3) | CD19 | Relapsed/refractory; Ph+ ALL; MRD+ |
| Inotuzumab ozogamicin | Antibody-drug conjugate | CD22 | Relapsed/refractory B-ALL |
| Tisagenlecleucel / Axicabtagene | CAR-T cells | CD19 | Relapsed/refractory B-ALL |
| Rituximab | Anti-CD20 mAb | CD20 | CD20+ B-ALL front-line; Burkitt |
| TKIs (dasatinib, ponatinib) | Kinase inhibitors | BCR-ABL1 | Ph+ ALL |
Prognosis
Cure rates by age:
| Population | Approximate OS |
|---|---|
| Children (standard risk) | ~90% |
| AYA (15-35 years) - pediatric-inspired | ≥70-80% |
| Adults 45-55 years | 50-60% |
| Adults 55-65 years | 30-40% |
| Elderly/frail patients | <10% |
| Burkitt leukaemia (+ rituximab, no SCT) | 80-90% |
Favourable prognostic features:
- Age 1-10 years, WBC < 50 × 10⁹/L at diagnosis
- Hyperdiploidy (>50 chromosomes)
- t(12;21) (ETV6-RUNX1) in children
- CALLA-positive (CD10+) immunophenotype
- Rapid early response / early achievement of MRD negativity
Unfavourable prognostic features:
- Age <1 or >10 years (in children); all adult ALL
- WBC > 50 × 10⁹/L at diagnosis
- t(9;22) Ph+ without TKI therapy
- t(4;11) KMT2A rearrangement
- Hypodiploidy (<44 chromosomes)
- Ph-like ALL
- T-ALL with early T-cell precursor (ETP-ALL) phenotype
- CNS involvement at diagnosis
- Slow response to induction; failure to achieve MRD negativity
Key Points Summary
- ALL is the most common childhood cancer - peak at 3-4 years, B-cell type predominantly
- T-ALL predominantly affects adolescent males and often presents with a mediastinal mass
- Cytogenetics and molecular biology (not just morphology/immunophenotype) drive modern classification and risk stratification
- The Philadelphia chromosome (BCR-ABL1) is the single most important cytogenetic abnormality to identify in adult ALL - it is found in ~25% and is treatable with TKIs
- Treatment follows induction → consolidation → maintenance, with CNS prophylaxis throughout
- Blinatumomab, inotuzumab, and CAR-T cells have transformed the management of relapsed/refractory ALL
- MRD monitoring (minimal residual disease) guides treatment decisions and predicts relapse
Sources:
- Harrison's Principles of Internal Medicine, 22nd Ed. (2025), Chapter 111: Acute Lymphoid Leukemia
- Goldman-Cecil Medicine, International Ed., Chapter 168: Acute Lymphoblastic Leukemia
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13: Lymphoid Neoplasms
Make 20 mcqs of with and explain the wrong one with gab between question and answer
Here are 20 MCQs on Acute Lymphoid Leukaemia (ALL), with answers and explanations of the wrong options clearly separated.
MCQs on Acute Lymphoid Leukaemia (ALL)
Q1. ALL is most commonly seen in which age group?
A) Neonates (< 1 month)
B) Children aged 3-4 years
C) Middle-aged adults (40-50 years)
D) Elderly (> 70 years)
✅ Answer: B) Children aged 3-4 years
❌ A - Neonates: Neonatal ALL is extremely rare. Infants < 1 year actually carry a poor prognosis when ALL does occur (KMT2A rearrangements common), but this is not the peak incidence age.
❌ C - Middle-aged adults: ALL incidence actually decreases through adulthood (30s-50s). It is predominantly a childhood disease.
❌ D - Elderly: While incidence rises again slightly in the elderly (and Ph+ ALL predominates there), the peak incidence is firmly in early childhood at 3-4 years.
Q2. Which of the following is the most common type of ALL?
A) T-cell ALL
B) Mature B-cell ALL (Burkitt type)
C) B-cell precursor ALL
D) Early T-cell precursor (ETP-ALL)
✅ Answer: C) B-cell precursor ALL
❌ A - T-cell ALL: T-ALL accounts for only ~15-25% of all ALL cases. It predominates in adolescent males and often presents as a thymic mass.
❌ B - Mature B-cell (Burkitt) ALL: This is the least common subtype, comprising only 3-5% of adult ALL and characterised by surface IgM expression and t(8;14).
❌ D - ETP-ALL: This is a specific high-risk subtype of T-ALL and represents a minority of ALL overall. It carries a poor prognosis.
Q3. The Philadelphia chromosome (BCR-ABL1) in ALL produces which fusion protein?
A) 210 kD (same as CML)
B) 190 kD (smaller than in CML)
C) 230 kD (larger than in CML)
D) 170 kD
✅ Answer: B) 190 kD (smaller than in CML)
❌ A - 210 kD: This is the fusion protein size characteristic of CML, not ALL. The difference in BCR breakpoint determines the protein size - ALL uses a more upstream breakpoint (minor BCR breakpoint), producing the smaller p190 protein.
❌ C - 230 kD: The p230 protein is associated with chronic neutrophilic leukaemia, an extremely rare CML variant.
❌ D - 170 kD: This is not a recognised BCR-ABL1 fusion protein size in either ALL or CML.
Q4. Which cytogenetic finding in childhood B-ALL carries the BEST prognosis?
A) Hypodiploidy (< 44 chromosomes)
B) t(4;11) KMT2A rearrangement
C) t(12;21) ETV6-RUNX1 fusion
D) t(9;22) Philadelphia chromosome
✅ Answer: C) t(12;21) ETV6-RUNX1 fusion
❌ A - Hypodiploidy: This is associated with a poor prognosis in B-ALL. Fewer chromosomes correlates with worse outcomes, in contrast to hyperdiploidy (>50 chromosomes), which is favourable.
❌ B - t(4;11): KMT2A-AF4 rearrangement is found in ~7% of adult ALL and carries a poor prognosis. It is also the hallmark of infant ALL (< 1 year), which has the worst outcome.
❌ D - t(9;22): The Philadelphia chromosome confers a poor prognosis when treated with chemotherapy alone. It has improved dramatically with TKI addition, but is still considered high-risk.
Q5. Which of the following is the hallmark surface antigen of "common ALL" (CALLA)?
A) CD7
B) CD10
C) CD20
D) CD3
✅ Answer: B) CD10
❌ A - CD7: CD7 is a T-cell antigen. All T-ALL cases express CD7. It is not a marker of common (B-precursor) ALL.
❌ C - CD20: CD20 is expressed in only 30-40% of B-precursor ALL and 86-100% of Burkitt/mature B-ALL. It is not the defining marker of common ALL. Its presence is important because it makes rituximab applicable.
❌ D - CD3: CD3 (surface or cytoplasmic) is the definitive T-cell marker. Surface CD3 defines mature T-ALL. It has no role in identifying B-lineage ALL.
Q6. T-ALL most commonly presents in which demographic?
A) Female infants under 1 year
B) Elderly women over 65 years
C) Adolescent males
D) Middle-aged females
✅ Answer: C) Adolescent males
❌ A - Female infants: Infant ALL is almost exclusively B-ALL with KMT2A rearrangements. T-ALL in infants is rare. There is no female predominance in T-ALL.
❌ B - Elderly women: The elderly ALL surge is predominantly Ph+ B-lineage ALL, not T-ALL. T-ALL incidence is highest in adolescence when the thymus is at maximum size.
❌ D - Middle-aged females: T-ALL has no female predominance and does not peak in middle age. Its peak mirrors the thymus's maximum functional size, which is in adolescence.
Q7. A patient with T-ALL presents with breathlessness and facial oedema. What is the most likely finding on chest imaging?
A) Pleural effusion
B) Anterior mediastinal mass
C) Bilateral hilar lymphadenopathy
D) Consolidation in the right lower lobe
✅ Answer: B) Anterior mediastinal mass
❌ A - Pleural effusion: While pleural effusions can occur secondary to a mediastinal mass in T-ALL (SVC obstruction, lymphatic obstruction), the primary and most characteristic finding is the thymic/mediastinal mass itself.
❌ C - Bilateral hilar lymphadenopathy: This pattern is classic for sarcoidosis or lymphoma (especially Hodgkin disease). T-ALL characteristically produces an anterior mediastinal mass from thymic origin.
❌ D - Right lower lobe consolidation: This is a pattern of infection or aspiration pneumonia - unrelated to ALL infiltration. T-ALL does not preferentially involve pulmonary parenchyma.
Q8. Which mutation is found in 50-70% of T-ALL cases?
A) BCR-ABL1
B) KMT2A rearrangement
C) NOTCH1 mutation
D) MYC translocation
✅ Answer: C) NOTCH1 mutation
❌ A - BCR-ABL1: This is the hallmark of Ph+ ALL, which is overwhelmingly a B-cell ALL finding. BCR-ABL1 is found in T-ALL only very rarely.
❌ B - KMT2A rearrangement: KMT2A (MLL) rearrangements are most common in infant B-ALL and adult ALL with t(4;11). They are not characteristic of T-ALL.
❌ D - MYC translocation t(8;14): MYC translocations are the hallmark of Burkitt lymphoma/leukaemia (mature B-ALL). They are not typical of T-ALL.
Q9. Complete remission (CR) is achieved after induction chemotherapy in what percentage of adults with ALL?
A) 40-50%
B) 60-70%
C) 80-90%
D) 95-100%
✅ Answer: C) 80-90%
❌ A - 40-50%: This figure would represent a very poor induction response, seen only in heavily pre-treated relapsed/refractory disease or highly resistant subtypes. Standard induction achieves far higher CR rates.
❌ B - 60-70%: This underestimates the efficacy of modern induction regimens. Even older regimens achieved >75% CR rates in adults.
❌ D - 95-100%: This is closer to the paediatric CR rate (~90%+) or the remarkable CR rate seen with blinatumomab + dasatinib in Ph+ ALL (98%). For adults overall, 80-90% is the correct figure.
Q10. Which of the following is the standard maintenance regimen in ALL?
A) Cyclophosphamide + doxorubicin for 6 months
B) 6-mercaptopurine (daily) + methotrexate (weekly)
C) High-dose cytarabine for 2 years
D) Imatinib monotherapy for 1 year
✅ Answer: B) 6-mercaptopurine (daily) + methotrexate (weekly)
❌ A - Cyclophosphamide + doxorubicin: These are induction/consolidation agents, not maintenance agents. Their toxicity profile (myelosuppression, cardiotoxicity) makes them unsuitable for prolonged low-dose outpatient use.
❌ C - High-dose cytarabine for 2 years: High-dose cytarabine is used in consolidation cycles. Prolonged high-dose cytarabine is neither safe nor practical as maintenance due to serious neurotoxicity and myelosuppression.
❌ D - Imatinib monotherapy for 1 year: A TKI is added to maintenance in Ph+ ALL specifically, but it is given alongside 6-MP/methotrexate, not as monotherapy. Duration is 2-2.5 years, not 1 year.
Q11. "Ph-like ALL" is important to recognise because:
A) It is cured by standard chemotherapy alone
B) It has kinase-activating mutations targetable by TKIs
C) It exclusively affects children under 5 years
D) It carries a favourable prognosis
✅ Answer: B) It has kinase-activating mutations targetable by TKIs
❌ A - Cured by standard chemotherapy alone: Ph-like ALL actually has a poor response to standard chemotherapy - this is precisely why recognising it matters. These patients need additional targeted therapy.
❌ C - Exclusively affects children under 5: Ph-like ALL accounts for 20-25% of adult ALL and is actually more common with increasing age. It is not confined to young children.
❌ D - Favourable prognosis: Ph-like ALL carries a poor prognosis when treated with chemotherapy alone. It was first identified as an unexplained poor-outcome group among BCR-ABL1-negative ALL.
Q12. Which of the following is the correct definition of CNS involvement in ALL?
A) Any headache in a patient with ALL
B) ≥5 cells/μL or leukemic blasts on CSF morphology
C) Cranial nerve palsy alone
D) Brain MRI showing leptomeningeal enhancement
✅ Answer: B) ≥5 cells/μL or leukemic blasts on CSF morphology
❌ A - Any headache: Headache is a symptom suggestive of CNS involvement but is completely non-specific. It can result from anaemia, infection, or other causes. Diagnosis requires CSF analysis.
❌ C - Cranial nerve palsy alone: While cranial nerve palsies can occur with CNS leukaemia, the formal diagnostic criterion is CSF-based (cell count + morphology), not clinical signs alone.
❌ D - MRI enhancement alone: MRI findings can support the diagnosis but the standard diagnostic definition used in ALL trials is the CSF criterion (≥5 WBC/μL or leukemic blasts on cytospin). MRI is not the defining criterion.
Q13. Blinatumomab works by:
A) Inhibiting BCR-ABL1 kinase activity
B) Linking CD3 on T cells to CD19 on B-ALL blasts
C) Delivering a cytotoxin to CD22+ cells
D) Blocking PD-1 checkpoint signalling
✅ Answer: B) Linking CD3 on T cells to CD19 on B-ALL blasts
❌ A - Inhibiting BCR-ABL1 kinase: This is the mechanism of TKIs (imatinib, dasatinib, ponatinib). Blinatumomab is a bispecific T-cell engager (BiTE) antibody, not a kinase inhibitor.
❌ C - Delivering a cytotoxin to CD22+ cells: This describes inotuzumab ozogamicin, a CD22-targeted antibody-drug conjugate (ADC) that delivers calicheamicin to CD22+ blasts. Blinatumomab does not carry a cytotoxic payload.
❌ D - Blocking PD-1: PD-1 inhibitors (pembrolizumab, nivolumab) are checkpoint inhibitors. Blinatumomab is a bispecific antibody that activates the patient's own T cells to kill CD19+ leukemic blasts - a different mechanism entirely.
Q14. Which congenital syndrome carries a 20-fold increased risk of developing leukaemia?
A) Turner syndrome
B) Marfan syndrome
C) Down syndrome (Trisomy 21)
D) Noonan syndrome
✅ Answer: C) Down syndrome (Trisomy 21)
❌ A - Turner syndrome (45,X): Turner syndrome is associated with cardiovascular malformations and gonadal dysgenesis but does NOT carry a significantly elevated leukaemia risk compared to the general population.
❌ B - Marfan syndrome: Marfan syndrome is a connective tissue disorder caused by FBN1 mutations. It is not associated with increased leukaemia risk. It carries risks of aortic dissection and lens dislocation.
❌ D - Noonan syndrome: Noonan syndrome (RAS pathway mutations) carries a modestly increased risk of juvenile myelomonocytic leukaemia (JMML), but NOT a 20-fold increased ALL risk. The 20-fold figure is specific to Down syndrome.
Q15. Which of the following best describes the duration of maintenance therapy in ALL (non-Burkitt)?
A) 6 months
B) 1 year
C) 2-2.5 years
D) 5 years
✅ Answer: C) 2-2.5 years
❌ A - 6 months: This is specifically the maintenance duration for Burkitt leukaemia (mature B-ALL), which requires only a short intensive course. It is not correct for standard B-ALL or T-ALL.
❌ B - 1 year: One year is insufficient to prevent relapse in standard ALL. Studies consistently show that 2-2.5 years of maintenance significantly reduces relapse rates compared to shorter durations.
❌ D - 5 years: Five-year maintenance was used in historical protocols and is excessive. Modern trials established that 2-2.5 years is optimal - longer duration adds toxicity (growth retardation, hepatotoxicity, infections) without additional benefit.
Q16. Bone pain in a child with ALL is caused by:
A) Pathological fractures through lytic lesions
B) Leukemic infiltration of the periosteum or expansion of the medullary cavity
C) Avascular necrosis from steroid use
D) Vitamin D deficiency from poor nutrition
✅ Answer: B) Leukemic infiltration of the periosteum or expansion of the medullary cavity
❌ A - Pathological fractures: While osteopenia and fractures can complicate ALL (especially with steroid therapy), the initial presenting bone pain is caused by leukemic infiltration and marrow expansion, not pathological fracture. Fractures are a complication, not the primary cause of bone pain.
❌ C - Avascular necrosis (AVN) from steroids: AVN is a recognised treatment complication of high-dose corticosteroids. However, bone pain at initial presentation - before any therapy has begun - cannot be attributed to steroid use.
❌ D - Vitamin D deficiency: ALL-associated bone pain is mechanical/infiltrative, not metabolic. Vitamin D deficiency causes osteomalacia, which has a very different clinical picture (proximal muscle weakness, diffuse bony tenderness, Looser's zones on X-ray).
Q17. In the induction regimen for ALL, what is the role of L-asparaginase?
A) It alkylates DNA to prevent blast cell replication
B) It depletes asparagine, which leukemic blasts cannot synthesise independently
C) It inhibits topoisomerase II, causing DNA strand breaks
D) It crosses the blood-brain barrier to treat CNS disease
✅ Answer: B) It depletes asparagine, which leukemic blasts cannot synthesise independently
❌ A - Alkylation of DNA: DNA alkylation is the mechanism of cyclophosphamide and other alkylating agents. L-asparaginase is an enzyme, not an alkylating agent - it works by substrate depletion, not DNA damage.
❌ C - Topoisomerase II inhibition: This is the mechanism of doxorubicin/daunorubicin (anthracyclines) and etoposide. L-asparaginase does not interact with topoisomerase.
❌ D - CNS penetration: L-asparaginase does NOT cross the blood-brain barrier. This is precisely why dedicated CNS prophylaxis (intrathecal methotrexate, high-dose systemic methotrexate) is needed. Dexamethasone is preferred over prednisone in induction partly because it does cross the blood-brain barrier.
Q18. "Leukemia cutis" in ALL refers to:
A) Painful ulceration of the oral mucosa
B) A raised, non-pruritic rash from leukemic skin infiltration
C) Purpuric rash due to thrombocytopenia
D) Drug-induced maculopapular eruption
✅ Answer: B) A raised, non-pruritic rash from leukemic skin infiltration
❌ A - Oral ulceration: Oral mucositis and ulcers are treatment complications (from methotrexate, cytarabine). They are not leukemia cutis. Gingival hypertrophy/bleeding is more typical of AML (especially M4/M5 monocytic types).
❌ C - Purpuric rash from thrombocytopenia: Thrombocytopenic purpura (petechiae/ecchymoses) is a flat, non-raised lesion caused by blood extravasation, not leukemic cell infiltration. Leukemia cutis is specifically a raised lesion representing actual leukemic cells in the dermis.
❌ D - Drug-induced eruption: Drug rashes are hypersensitivity reactions and are treatment-related, not disease-related. They may be maculopapular, urticarial, or morbilliform - distinct from the specific raised papules/nodules of leukemia cutis.
Q19. Which surface marker is a target of CAR-T cell therapy in B-ALL?
A) CD7
B) CD19
C) CD56
D) CD33
✅ Answer: B) CD19
❌ A - CD7: CD7 is a T-cell marker. While CD7-targeted CAR-T is under investigation for T-ALL, the established, approved CAR-T therapies in ALL (tisagenlecleucel, etc.) target CD19 on B-lineage blasts.
❌ C - CD56: CD56 (NCAM) is expressed on NK cells and plasma cells. It is a target under investigation in multiple myeloma (daratumumab indirectly relevant) but is not the primary CAR-T target in ALL.
❌ D - CD33: CD33 is expressed on myeloid cells. It is the target of gemtuzumab ozogamicin in AML, not ALL. Using CD33-targeted therapy in B-ALL would miss the target entirely, as B-lymphoblasts do not significantly express CD33.
Q20. Which of the following statements about the outcome of ALL is CORRECT?
A) Adults have better outcomes than children due to more aggressive chemotherapy
B) The major risk of relapse peaks at 5 years after remission
C) Burkitt leukaemia treated with intensive chemotherapy + rituximab achieves ~80-90% overall survival
D) Allogeneic SCT is recommended for all ALL patients in first remission
✅ Answer: C) Burkitt leukaemia treated with intensive chemotherapy + rituximab achieves ~80-90% overall survival
❌ A - Adults have better outcomes than children: The opposite is true. Children achieve ~90% cure rates while adult outcomes are strictly worse with age, falling to < 10% in elderly/frail patients. The improved adult outcomes with "pediatric-inspired" protocols actually comes from adopting children's regimens.
❌ B - Major relapse risk peaks at 5 years: The major risk of relapse is in the first 2 years after achieving remission. Relapse is much less likely after 5 years. This is why consolidation and maintenance therapy are most intensive in the first 2-2.5 years.
❌ D - Allogeneic SCT for ALL patients in first remission: SCT is reserved for high-risk patients in first CR (e.g., Ph+, KMT2A rearrangement, poor induction response, MRD+ after consolidation). It is not recommended for all patients - particularly standard-risk paediatric and adolescent patients do very well without SCT and its attendant morbidity/mortality.
Based on: Harrison's Principles of Internal Medicine 22nd Ed., Goldman-Cecil Medicine, Robbins & Cotran Pathologic Basis of Disease
Cml
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Excellent - rich content from Harrison's 22nd Ed and Robbins/Goldman-Cecil. I now have everything needed for a thorough CML overview.
Chronic Myeloid Leukaemia (CML)
Definition
CML is a clonal hematopoietic stem cell myeloproliferative neoplasm driven by the BCR-ABL1 chimeric oncogene, produced by a reciprocal translocation between chromosomes 9 and 22 - t(9;22)(q34.1;q11.2) - known as the Philadelphia (Ph) chromosome.
- The Ph chromosome = elongated chromosome 9 + shortened chromosome 22
- BCR-ABL1 encodes a constitutively active p210 tyrosine kinase (in CML)
- This kinase drives uncontrolled granulocytic proliferation via RAS, JAK/STAT, and PI3K/AKT signalling pathways
- There is no BCR-ABL1-negative CML - cases without the Ph chromosome by cytogenetics still harbour BCR-ABL1 detectable by FISH or PCR
Epidemiology
| Parameter | Data |
|---|---|
| % of all leukaemias | ~15% |
| Annual incidence | 2/100,000; ~9,000 new cases/year (USA) |
| Median age at diagnosis | 55-65 years |
| Male predominance | Male:Female ratio 1.6:1 |
| Children (<20 years) | Only ~3% of CML |
| Peak incidence | Fifth to sixth decade of life |
With TKI therapy, annual CML mortality has fallen from 10-20% to just ~1-2%, meaning the prevalence of CML is projected to plateau at ~450,000 in the USA by 2040 - making CML an increasingly common oncology diagnosis despite its relatively low incidence.
Etiology
- No familial association - no increased risk in monozygotic twins or relatives
- No benzene, insecticide, fertilizer, or viral associations (unlike AML)
- Ionizing radiation is the only established risk factor:
- Risk peaks 5-10 years after exposure
- Dose-related
- Mean time to CML development after atomic bomb exposure: 6.3 years
Pathophysiology
The t(9;22) translocation juxtaposes ABL1 (chromosome 9q34) to BCR (chromosome 22q11). Two main BCR breakpoints (e13 or e14) produce transcripts e13a2 and e14a2, both encoding the p210BCR-ABL1 oncoprotein.
Signalling consequences:
- Constitutive tyrosine kinase activity → phosphorylation of downstream substrates
- Activation of RAS pathway (proliferation)
- Activation of JAK/STAT pathway (survival, inhibited apoptosis)
- BCR-ABL1 dimerisation (via BCR coiled-coil domain) is required for kinase activation
- Preferentially drives granulocytic and megakaryocytic progenitor expansion
- Causes abnormal release of immature granulocytes from marrow into blood
- Normal stem cells persist and can re-emerge with effective TKI therapy
Compare BCR-ABL1 fusion proteins:
Protein Size Associated condition p210 210 kDa CML (most); rare in Ph+ ALL p190 190 kDa Ph+ ALL (2/3 of cases) p230 230 kDa Rare CML/chronic neutrophilic leukaemia
Phases of CML
CML follows a biphasic or triphasic course:
1. Chronic Phase (CP) - ~90% at diagnosis
- Symptomatic but manageable
- Blasts < 10% (WHO) or < 15% (ELN) in peripheral blood/marrow
- Duration: average 3-6 years without treatment
- >75% of CML in developed world diagnosed in this phase
2. Accelerated Phase (AP)
| Feature | ELN criteria | WHO criteria |
|---|---|---|
| Blast % | 15-30% | 10-20% |
| Additional cytogenetic abnormalities | Clonal evolution (e.g., trisomy 8, isochromosome 17q, duplication of Ph) | Similar |
| Other features | Basophils ≥20%, refractory thrombocytopenia | Similar |
Note: ELN criteria are used in clinical practice; WHO criteria are rarely used in trials.
3. Blast Phase (Blast Crisis) - BC
| Criterion | ELN | WHO |
|---|---|---|
| Blast % | ≥30% | ≥20% |
- 70% myeloid blast crisis (resembles AML)
- 30% lymphoid blast crisis (usually pre-B cell) - evidence CML originates from a pluripotent stem cell with both myeloid and lymphoid potential
- Blast crisis is triggered by acquisition of additional mutations in transcription factor genes (e.g., RUNX1, GATA2, WT1, TP53)
- Prognosis: median survival 5-7 months in blast crisis
Morphology
Peripheral Blood:
- Leukocytosis, often exceeding 100,000 cells/μL
- Full granulocytic spectrum: neutrophils, band forms, metamyelocytes, myelocytes
- Eosinophilia and basophilia (characteristic)
- Blasts usually < 10% in chronic phase
- Thrombocytosis (sometimes marked)
- Low Leukocyte Alkaline Phosphatase (LAP) score - key differentiator from leukaemoid reaction
Bone Marrow:
- Markedly hypercellular - massively increased maturing granulocytic precursors
- Increased megakaryocytes (small, dysplastic forms)
- Sea-blue histiocytes (scattered macrophages with wrinkled green-blue cytoplasm) - characteristic
- Increased reticulin; overt fibrosis rare in chronic phase
Spleen:
- Greatly enlarged due to extensive extramedullary haematopoiesis
- Contains infarcts of varying age
- Mild hepatomegaly and lymphadenopathy also possible
Clinical Features
Onset: insidious
- Fatigue, weakness, weight loss, anorexia (hypermetabolic state)
- Dragging sensation in the left upper abdomen from massive splenomegaly
- Acute left upper quadrant pain from splenic infarction
- Mild-to-moderate anaemia
- Symptoms of hyperviscosity at very high WBC counts
- ~25-50% diagnosed incidentally on routine FBC
Hepatosplenomegaly:
- Splenomegaly is the most prominent finding on examination
- Can be massive - extending to the pelvis in advanced disease
Diagnosis
Step 1 - FBC + Peripheral Blood Film
- Leukocytosis with left shift (entire granulocytic series)
- Basophilia + eosinophilia
- Low/absent LAP score
Step 2 - Confirm BCR-ABL1
| Test | Role |
|---|---|
| Cytogenetics (karyotype) | Detects Ph chromosome in >90%; also detects additional cytogenetic abnormalities (clonal evolution) |
| FISH | Detects BCR-ABL1 in Ph-negative CML; quantitative |
| RT-PCR (qPCR) | Gold standard for monitoring MRD (minimal residual disease); quantifies BCR-ABL1 transcript on International Scale (IS) |
Step 3 - Bone Marrow Biopsy
- Confirms phase, assesses fibrosis
- Cytogenetics for clonal evolution
- Not always required for diagnosis in classical CML
TKI Therapy - The Revolution
Before TKIs (pre-2001): median survival 3-7 years, 10-year survival ≤30%.
After TKIs: 10-year survival >85%, approaching normal age-matched population.
FDA-approved BCR-ABL1 TKIs:
| Generation | Agent | Dose | Key Toxicities |
|---|---|---|---|
| 1st | Imatinib (Gleevec) | 400 mg daily | Oedema (37%), muscle cramps (41%), hypophosphataemia (28%) |
| 2nd | Dasatinib (Sprycel) | 100 mg daily | Pleural/pericardial effusions (28%), neutropenia, pulmonary hypertension |
| 2nd | Nilotinib (Tasigna) | 300-400 mg BID | Rash (38%), QTc prolongation, hyperglycaemia, cardiovascular events |
| 2nd | Bosutinib (Bosulif) | 400 mg daily | Diarrhoea (70%), elevated ALT (23%), thrombocytopenia |
| 3rd | Ponatinib (Iclusig) | Variable | Arterial thrombosis, hypertension (inhibits VEGFR), pancreatitis |
| 3rd/STAMP | Asciminib (Scemblix) | Variable | Unique mechanism - targets ABL myristoyl pocket (STAMP inhibitor) |
Key potency comparisons vs imatinib:
- Nilotinib: ~30× more potent
- Bosutinib: 30-50× more potent
- Dasatinib: ~300× more potent; also inhibits SRC family kinases
Front-line Response Data (12-month milestones):
| Drug | CCyR at 12m | MMR at 12m |
|---|---|---|
| Imatinib | 65-66% | 22-37% |
| Nilotinib | 78-80% | 43-44% |
| Dasatinib | 83% | 46% |
| Bosutinib | 77% | 47% |
(CCyR = complete cytogenetic response; MMR = major molecular response = BCR-ABL1 ≤0.1% IS)
TKI Response Milestones (ELN 2020):
| Timepoint | Optimal | Warning | Failure |
|---|---|---|---|
| 3 months | BCR-ABL1 ≤10% IS | BCR-ABL1 >10% IS | No CHR or Ph+ >95% |
| 6 months | BCR-ABL1 <1% IS (CCyR) | BCR-ABL1 1-10% IS | BCR-ABL1 >10% or Ph+ >35% |
| 12 months | BCR-ABL1 ≤0.1% IS (MMR) | BCR-ABL1 0.1-1% IS | BCR-ABL1 >1% or Ph+ >0% |
Resistance & T315I Mutation
The T315I "gatekeeper" mutation is the most important resistance mutation:
- Threonine → Isoleucine substitution at position 315 of ABL1
- Confers resistance to imatinib, dasatinib, nilotinib, and bosutinib
- Only ponatinib and asciminib retain activity against T315I
- Asciminib targets the ABL myristoyl pocket (allosteric site) rather than the ATP-binding site - unique mechanism among TKIs
Treatment-Free Remission (TFR)
A major modern goal - stopping TKI therapy in patients who achieve deep molecular response:
- Requires ≥4.5-log reduction in BCR-ABL1 (MR4.5) sustained for ≥2 years
- ~50% of carefully selected patients maintain molecular remission after stopping TKI
- The other ~50% relapse (usually within 6 months) but re-respond to TKI re-initiation
- TFR eliminates lifelong drug costs and toxicities
Allogeneic SCT
- Was the only curative option pre-TKI era
- Now reserved for TKI failure (multiple TKIs) or blast crisis
- Risks include transplant-related mortality (10-30%), GvHD, infections
- Accelerated phase with TKIs: median survival now 88 months (vs 28 months pre-TKI)
- Blast crisis remains devastating: median survival only 5-7 months
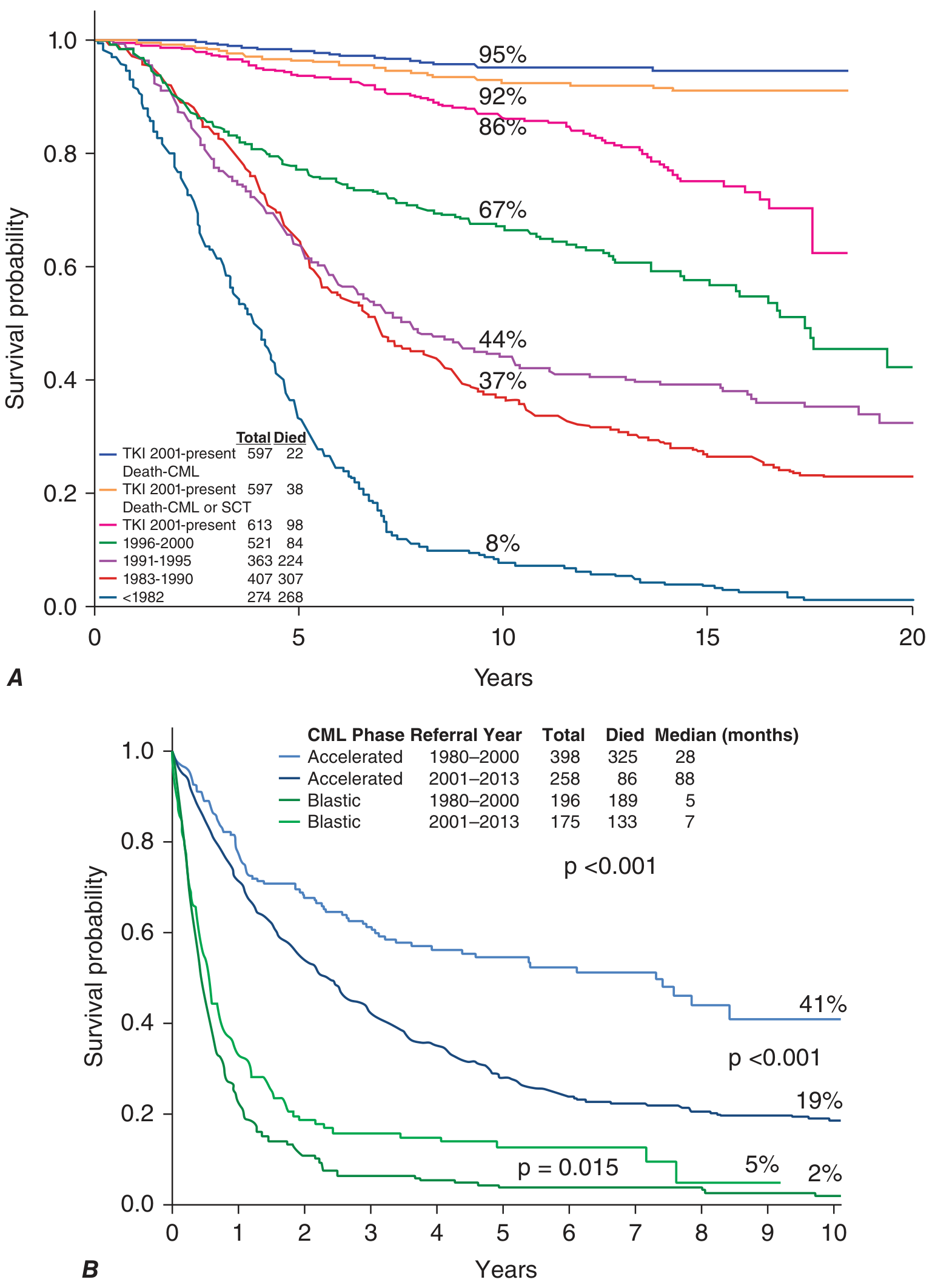
Sokal and ELTS Scoring - Risk Stratification at Diagnosis
Sokal score (older) and ELTS (EUTOS Long-Term Survival) score (preferred now) use:
- Age
- Spleen size
- Platelet count
- % blasts at diagnosis
These stratify patients into low, intermediate, and high risk - influencing choice of front-line TKI.
Key Differentials
| Feature | CML | Leukaemoid Reaction |
|---|---|---|
| LAP score | Low/absent | Very high |
| BCR-ABL1 | Present | Absent |
| Basophilia | Prominent | Absent/mild |
| Cause | Neoplastic | Infection/inflammation |
| Splenomegaly | Massive | Mild/absent |
Summary - Key Points
- CML is caused by t(9;22)(q34;q11) - the Philadelphia chromosome - present in >90% of cases; the rest have cryptic BCR-ABL1 detectable by FISH/PCR
- The cell of origin is a pluripotent HSC - explains why blast crisis can be myeloid OR lymphoid
- BCR-ABL1 produces p210 kDa protein in CML (vs p190 in Ph+ ALL)
- Disease course: chronic → accelerated → blast crisis (the latter carrying very poor prognosis)
- TKIs (especially imatinib and 2nd-gen TKIs) have transformed CML from a uniformly fatal disease to one with near-normal life expectancy
- T315I mutation = pan-resistance to 1st/2nd gen TKIs; only ponatinib/asciminib work
- MRD monitoring by qPCR (BCR-ABL1 on International Scale) drives all treatment decisions
- Treatment-free remission is now an achievable goal in ~50% of patients with deep molecular response
Sources: Harrison's Principles of Internal Medicine 22nd Ed. (2025), Chapter 110; Goldman-Cecil Medicine, Chapter 170; Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13
AML
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from Harrison's 22nd Ed and Goldman-Cecil. Let me compile the complete, detailed AML overview.
Acute Myeloid Leukaemia (AML)
Definition
AML is an aggressive clonal neoplasm of immature myeloid lineage blast cells (myeloblasts, monoblasts, erythroblasts, or megakaryoblasts) that replace the bone marrow and suppress normal haematopoiesis. It is characterised by:
- Arrest of myeloid differentiation at an early precursor stage
- Accumulation of non-functional blast cells
- Resultant bone marrow failure (anaemia, thrombocytopenia, neutropenia)
Diagnostic threshold: ≥20% blasts in bone marrow or peripheral blood (exceptions: AML with t(8;21), inv(16), or t(15;17) are diagnosed regardless of blast count given their defining genetic abnormalities)
Epidemiology
| Parameter | Data |
|---|---|
| Most common acute leukaemia in adults | Yes (ALL is more common in children) |
| Median age at diagnosis | ~68 years |
| Annual incidence (USA) | ~20,000 new cases/year |
| Male slight predominance | Yes |
| 5-year overall survival (all ages) | ~29% overall; ~40-50% in younger fit adults with favourable genetics |
Aetiology & Risk Factors
1. Acquired / Environmental
| Exposure | Comment |
|---|---|
| Alkylating agents (chlorambucil, cyclophosphamide, melphalan) | Therapy-related AML; latency 5-10 years; often complex karyotype / del(5q) / del(7q) |
| Topoisomerase II inhibitors (etoposide, doxorubicin) | Therapy-related AML; latency 1-3 years; often KMT2A (11q23) rearrangements |
| Benzene / petroleum products | Chronic occupational exposure |
| Ionizing radiation | Historical (radiologists, atomic bomb survivors) |
| Cigarette smoking | Associated, possibly via benzene metabolites |
| Prior MDS or MPN | Secondary AML - worse prognosis |
2. Hereditary / Germline Predisposition
| Syndrome | Mechanism |
|---|---|
| Down syndrome (Trisomy 21) | GATA1 mutation → megakaryoblastic AML (before age 4); excellent outcomes with dose-reduced chemo |
| Fanconi anaemia | Defective DNA repair → AML |
| Bloom syndrome | Defective DNA repair |
| Ataxia-telangiectasia | Defective DNA repair |
| Kostmann syndrome (congenital neutropenia) | G-CSF receptor + ELANE mutations → AML |
| Shwachman-Diamond syndrome | Ribosome assembly defect |
| Li-Fraumeni syndrome | Germline TP53 mutation |
| Familial AML with DDX41 | Germline DDX41 - increasingly recognised |
Anticancer drugs are the leading cause of therapy-related AML.
Pathogenesis
AML results from two complementary categories of mutations ("two-hit" model):
| Class | Examples | Effect |
|---|---|---|
| Class I - activate proliferation/survival | FLT3-ITD, RAS mutations, KIT mutations | Growth advantage, anti-apoptosis |
| Class II - impair differentiation | RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA, CEBPA, NPM1 | Maturation arrest at blast stage |
AML cells carry on average 10-15 mutations per cell (~5 driver mutations), far fewer than solid tumours.
Key driver mutations and their frequency:
| Gene | Frequency | Prognostic Impact |
|---|---|---|
| FLT3-ITD | 30-35% | Adverse (especially high allelic ratio) |
| NPM1 | ~30% | Favourable (if no FLT3-ITD) |
| DNMT3A | ~20% | Intermediate-adverse |
| IDH1 | ~8% | Targetable (ivosidenib) |
| IDH2 | ~12% | Targetable (enasidenib) |
| CEBPA (bZIP biallelic) | 4-15% | Favourable |
| TP53 | ~8-15% | Strongly adverse |
| RUNX1 | ~10% | Adverse |
| ASXL1 | ~10% | Adverse |
| KIT | ~5-10% | In CBF-AML |
WHO 2022 Classification
Group 1: AML with Defining Genetic Abnormalities
(diagnosed regardless of blast count if genetic lesion is present)
| Subtype | Genetic Lesion | Notes |
|---|---|---|
| APL | PML::RARA fusion; t(15;17) | Medical emergency; ATRA + ATO treatment |
| CBF-AML | RUNX1::RUNX1T1 fusion; t(8;21) | Favourable; slender Auer rods; CD19+ |
| CBF-AML | CBFB::MYH11; inv(16)/t(16;16) | Favourable; abnormal eosinophils in marrow |
| DEK::NUP214; t(6;9) | Adverse | |
| RBM15::MRTFA; t(1;22) | Megakaryoblastic; infants | |
| BCR::ABL1 fusion* | Rare | |
| KMT2A rearrangement | Adverse (most partners); monocytic features | |
| MECOM rearrangement | Adverse | |
| NUP98 rearrangement | Adverse | |
| NPM1 mutation | Most common (~30%); favourable if no FLT3-ITD | |
| CEBPA bZIP mutation | Favourable |
Group 2: AML, Myelodysplasia-Related (AML-MR)
- Prior MDS or MDS/MPN history, OR
- Complex karyotype, specific chromosomal aberrations, OR
- Mutations in: ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2
- Carries adverse prognosis
Group 3: AML, Defined by Differentiation (NOS)
(when no defining genetic abnormality found)
- AML with minimal differentiation (M0)
- AML without maturation (M1)
- AML with maturation (M2)
- Acute myelomonocytic leukaemia (M4)
- Acute monocytic leukaemia (M5)
- Acute erythroid leukaemia (M6)
- Acute megakaryoblastic leukaemia (M7)
- Acute basophilic leukaemia
Note: The 2022 WHO classification superseded the 2016 version. The older FAB (French-American-British) M0-M7 classification is now largely replaced but still used in some contexts.
Morphology & Special Features
Auer Rods
- Pathognomonic of AML (never seen in ALL)
- Needle-shaped pink cytoplasmic inclusions = fused lysosomes
- Multiple Auer rods in a single cell = "faggot cells" - hallmark of APL (M3)
Key Morphologic-Genetic Correlations:
| Morphologic Feature | Associated Genetics |
|---|---|
| Faggot cells (multiple Auer rods) | APL / t(15;17) / PML-RARA |
| Slender Auer rods + CD19 expression + normal eosinophils | t(8;21) / RUNX1-RUNX1T1 |
| Abnormal marrow eosinophils | inv(16) / CBFB-MYH11 |
| Cup-shaped nuclear blasts | NPM1 mutation (especially + FLT3) |
| Monocytic features + gingival hypertrophy | KMT2A rearrangements; AML M5 |
| Dysplastic morphology | Complex karyotype / TP53 mutation |
| DIC at presentation | APL / t(15;17) |
Immunophenotype (Flow Cytometry):
| Marker | Expression Pattern |
|---|---|
| CD34, CD117, HLA-DR | Most immature AML forms |
| CD13, CD33 | More differentiated myeloid AML |
| CD14, CD15, CD11b | Monocytic AML |
| CD36, CD71, CD235a (glycophorin A) | Erythroid leukaemia |
| CD41, CD61 | Megakaryoblastic AML |
Clinical Features
From Marrow Failure:
- Anaemia - fatigue, dyspnoea, decreased exercise tolerance
- Thrombocytopenia - petechiae, ecchymoses, bleeding from unusual sites, DIC (especially APL)
- Neutropenia - recurrent or life-threatening bacterial/fungal infections; fever is the most common presenting symptom
From Organ Infiltration:
- Gingival hypertrophy - characteristic of monocytic AML (M5) / KMT2A rearrangements
- Leukemia cutis (skin nodules/rash) - most common in monocytic subtypes
- Myeloid sarcoma (chloroma) - extramedullary tumour of myeloid blasts; green colour from myeloperoxidase; associated with t(8;21)
- Hepatosplenomegaly, lymphadenopathy (less prominent than in ALL)
- CNS involvement - headache, visual changes, cranial nerve palsies (rare in AML vs ALL)
- Back pain + lower extremity weakness - spinal granulocytic sarcoma; associated with t(8;21)
- DIC - characteristic of APL; ecchymoses, oozing from IV sites
Hyperleukocytosis (WBC > 100,000/μL):
- Occurs in ~5-10% of AML
- Leukostasis: slugging of blasts in capillaries → respiratory failure, stroke, priapism
- Medical emergency - requires emergent leukapheresis
Diagnosis
Initial Workup:
- CBC with differential - anaemia, thrombocytopenia, leukocytosis (or leukopenia); blasts on smear
- Bone marrow aspirate + biopsy (essential):
- Morphology (Auer rods, blast %)
- Flow cytometry (immunophenotype)
- Cytogenetics (karyotype) - standard
- FISH - specific translocations
- Molecular studies (NGS panel): NPM1, FLT3, IDH1/2, CEBPA, TP53, ASXL1, DNMT3A, etc.
- Chemistry panel: LDH, uric acid (tumour lysis risk), electrolytes, renal/hepatic function, DIC screen (PT, aPTT, fibrinogen, d-dimer)
- Cardiac function: echocardiogram or MUGA scan (before anthracyclines)
- HLA typing: for potential allogeneic SCT
- Viral serologies: CMV, HSV, VZV (reactivation risk)
ELN 2022 Risk Stratification (Crucial for Treatment Decisions)
Favourable Risk:
- t(8;21)/RUNX1::RUNX1T1
- inv(16) or t(16;16)/CBFB::MYH11
- Mutated NPM1 without FLT3-ITD
- bZIP in-frame mutated CEBPA
Intermediate Risk:
- Mutated NPM1 with FLT3-ITD
- Wild-type NPM1 with FLT3-ITD (no adverse lesions)
- t(9;11)/MLLT3::KMT2A
- Other cytogenetic/molecular abnormalities
Adverse Risk:
- t(6;9)/DEK::NUP214
- t(v;11)/KMT2A-rearranged (most partners)
- t(9;22)/BCR::ABL1
- inv(3) or t(3;3)/GATA2,MECOM(EVI1)
- -5 or del(5q); -7; -17/abnl(17p)
- Complex karyotype (≥3 abnormalities)
- Monosomal karyotype (≥2 monosomies or 1 monosomy + structural abnormality)
- Wild-type NPM1 + FLT3-ITD high
- Mutated RUNX1, ASXL1, TP53
Treatment
Overall Framework:
Treatment is divided into Induction → Consolidation (Post-remission therapy), selected based on:
- Patient age and fitness
- ELN genetic risk category
- Availability of targeted agents
Induction Chemotherapy
Standard-intensity ("7+3 regimen") - for fit patients:
- Cytarabine 100-200 mg/m² continuous IV infusion × 7 days
- Anthracycline (daunorubicin 60-90 mg/m² or idarubicin 12 mg/m²) × days 1, 2, 3
- CR rate: 60-80% in younger patients; lower in older/unfit
Mechanism of key drugs:
- Cytarabine: S-phase specific antimetabolite → inhibits DNA synthesis (phosphorylated to ara-CTP intracellularly)
- Anthracyclines: DNA intercalators → topoisomerase II inhibition → DNA strand breaks
Addition to 7+3:
- Gemtuzumab ozogamicin (anti-CD33 ADC) added in CBF-AML and CD33+ AML
- Midostaurin added for FLT3-mutated AML (first FDA approval in AML with a targeted agent)
- Gilteritinib (FLT3 inhibitor) for relapsed/refractory FLT3-mutated AML
Low-intensity regimens - for older/unfit patients:
- Venetoclax + azacitidine (or decitabine) - now standard of care for unfit elderly patients
- Venetoclax = BCL-2 inhibitor → overcomes apoptosis resistance
- CR rates ~65-70% in previously untreated elderly patients
- IDH1 inhibitor (ivosidenib) ± azacitidine - for IDH1-mutated AML
- IDH2 inhibitor (enasidenib) - for IDH2-mutated AML
- Low-dose cytarabine (LDAC) ± glasdegib
Post-remission / Consolidation
After achieving CR, further therapy is mandatory (relapse is near-universal without it):
| Risk Category | Preferred Post-remission Strategy |
|---|---|
| Favourable (CBF-AML, NPM1 without FLT3) | High-dose cytarabine (HiDAC) × 3-4 cycles; no SCT needed in first CR |
| Intermediate | HiDAC consolidation or allogeneic SCT - individualised based on MRD |
| Adverse | Allogeneic SCT in first CR is standard |
| Therapy-related / AML-MR | Allogeneic SCT if possible |
High-dose cytarabine (HiDAC): 1-3 g/m² given 4-12 doses per course
- Key toxicity: cerebellar toxicity (ataxia, dysarthria) - monitor carefully
Acute Promyelocytic Leukaemia (APL) - Special Case
APL (AML with PML::RARA / t(15;17)) is a medical emergency but also has the best prognosis of all AML subtypes.
Pathophysiology: PML-RARA fusion protein blocks myeloid differentiation at the promyelocytic stage and releases granule contents → DIC.
Treatment:
- ATRA (all-trans retinoic acid) + ATO (arsenic trioxide) = standard of care for low-risk APL
- ATRA: 45 mg/m²/day orally → induces differentiation of leukemic promyelocytes
- ATO: 0.15 mg/kg/day IV
- CR approaches 100%; long-term survival >90%
- High-risk APL (WBC >10,000/μL): add cytoreductive chemotherapy immediately due to risk of APL syndrome and DIC
APL (Differentiation) Syndrome:
- Occurs within first 3 weeks of ATRA/ATO therapy
- Features: fever, fluid retention, dyspnoea, chest pain, pulmonary infiltrates, pleural/pericardial effusions, hypoxaemia
- Mechanism: adhesion of differentiated neoplastic cells to pulmonary vasculature
- Treatment: dexamethasone ± cytoreduction; temporarily stop ATRA if severe
- Mortality: ~10% if unrecognised
MRD monitoring in APL: PCR for PML-RARA - disappearance of signal = long-term DFS; reemergence = relapse.
Targeted Agents in AML (Summary)
| Drug | Target | Indication |
|---|---|---|
| Midostaurin | FLT3 inhibitor (multi-kinase) | Frontline + 7+3 for FLT3-mutated AML |
| Gilteritinib | FLT3 inhibitor (selective) | Relapsed/refractory FLT3-mutated AML |
| Quizartinib | FLT3 inhibitor | FLT3-ITD AML |
| Ivosidenib | IDH1 inhibitor | IDH1-mutated AML (frontline or R/R) |
| Enasidenib | IDH2 inhibitor | IDH2-mutated AML (R/R) |
| Venetoclax | BCL-2 inhibitor | + azacitidine for unfit elderly AML |
| Gemtuzumab ozogamicin | Anti-CD33 ADC | CBF-AML + 7+3; CD33+ AML |
| ATRA | RAR-α ligand → differentiation | APL (PML-RARA) |
| Arsenic trioxide | Degrades PML-RARA fusion | APL |
| Revumenib | Menin inhibitor | KMT2A-rearranged or NPM1-mutated AML (R/R) |
Allogeneic SCT in AML
- Gold standard for adverse-risk AML in first CR
- Only potentially curative approach for many high-risk patients
- Requires HLA-matched sibling or unrelated donor (or haploidentical if no match)
- Main benefit: Graft-vs-Leukaemia (GvL) effect in addition to high-dose conditioning
- Main risks: transplant-related mortality, graft-vs-host disease (GvHD), infections
- Decision guided by ELN risk + MRD status after induction
MRD (Minimal Residual Disease) Monitoring
MRD assessment after induction and consolidation is increasingly standard:
- Flow cytometry MRD (leukemia-associated immunophenotype)
- Molecular MRD (NPM1, RUNX1-RUNX1T1, CBFB-MYH11 by PCR)
- MRD negativity after first consolidation = strong predictor of long-term remission
- MRD persistence → escalate therapy / proceed to SCT
Prognosis
| Risk Group | 5-year OS (approximate) |
|---|---|
| Favourable (CBF-AML, NPM1+/FLT3-) | ~50-70% |
| Intermediate | ~25-40% |
| Adverse (complex karyotype, TP53, monosomal) | <10-15% |
| APL (low-risk, ATRA + ATO) | >90% |
| Therapy-related AML | ~10-20% |
| Elderly patients overall | ~10-20% |
Key Points Summary
- AML is diagnosed by ≥20% blasts in marrow/blood (except APL, t(8;21), inv(16) - diagnosed by genetics)
- Auer rods are pathognomonic of AML; faggot cells (multiple Auer rods) = APL
- The two-hit model: Class I mutations (FLT3, RAS) drive proliferation; Class II mutations (NPM1, RUNX1 fusions) block differentiation
- APL is a haematological emergency - DIC is the key early threat; ATRA + ATO achieves ~100% CR and >90% long-term survival
- FLT3-ITD (~30%) = most common adverse mutation; NPM1 (~30%) = most common favourable mutation
- Treatment: 7+3 induction → HiDAC consolidation for fit patients; venetoclax + azacitidine for unfit elderly patients
- ELN risk stratification determines whether allogeneic SCT is needed in first CR
- Targeted therapies (midostaurin, gilteritinib, ivosidenib, venetoclax) have significantly improved outcomes across AML subtypes
Sources: Harrison's Principles of Internal Medicine 22nd Ed. (2025), Chapter 109; Goldman-Cecil Medicine, Chapter 168; Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13
Make 40 mcqs pf aml clinical based scenarios along with explanation of answers and wrong ones covering every mahor aspect of topic at final year mbbs level with gap between question and answer
This is a shared conversation. Sign in to Orris to start your own chat.