
Screenshot_20260621-123802.png
Explain
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough explanation. Here is the full breakdown:
Pancreatic Neuroendocrine Neoplasms (PanNENs)
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 1011-1012
Overview
The preferred term for what were historically called "islet cell tumors" is pancreatic neuroendocrine neoplasms (PanNENs). They are rare, accounting for only 2% of all pancreatic neoplasms, but they are clinically important because many secrete hormones that produce dramatic clinical syndromes.
Key general features:
- Can occur anywhere in the pancreas or adjacent peripancreatic tissues
- May be single or multiple, and of varying histologic grades
- Graded by mitotic activity and Ki-67 proliferative index
- Poorly differentiated neuroendocrine carcinomas (small cell or large cell type) have Ki-67 >20%
- May be functional (hormone-secreting) or nonfunctional
- Prognosis: ~90% of insulin-producing tumors are benign; 60-90% of other subtypes are malignant
Pathogenesis involves recurrent mutations in three major pathways:
- MEN1 gene (also causes familial MEN syndrome type 1)
- PTEN/TSC2 loss-of-function → activates mTOR signaling
- ATRX/DAXX inactivation → alternative lengthening of telomeres (ALT); found in ~50% of PanNENs
1. Hyperinsulinism (Insulinoma) - p. 1011
Insulinomas are the most common PanNEN subtype and arise from beta (β) cells of the islets of Langerhans.
Pathophysiology
The tumor secretes insulin autonomously, independent of blood glucose levels, causing hypoglycemia.
Clinical Features - Whipple's Triad (classic)
- Hypoglycemic episodes when blood glucose falls below 50 mg/dL
- Symptoms: confusion, stupor, loss of consciousness (neurological due to glucose deprivation)
- Episodes precipitated by fasting or exercise
- Promptly relieved by feeding or IV glucose
Key Lab Finding
- Inappropriately high circulating insulin in the setting of hypoglycemia
- Elevated C-peptide confirms endogenous (not exogenous) hyperinsulinism
Morphology
- Usually solitary, small (<2 cm), encapsulated, pale-to-red-brown nodules
- Look like giant islets histologically - monotonous cells, minimal pleomorphism, minimal mitotic activity
- Only ~10% are malignant (carcinomas)
- Characteristic histologic feature: amyloid deposition (pink material on H&E)
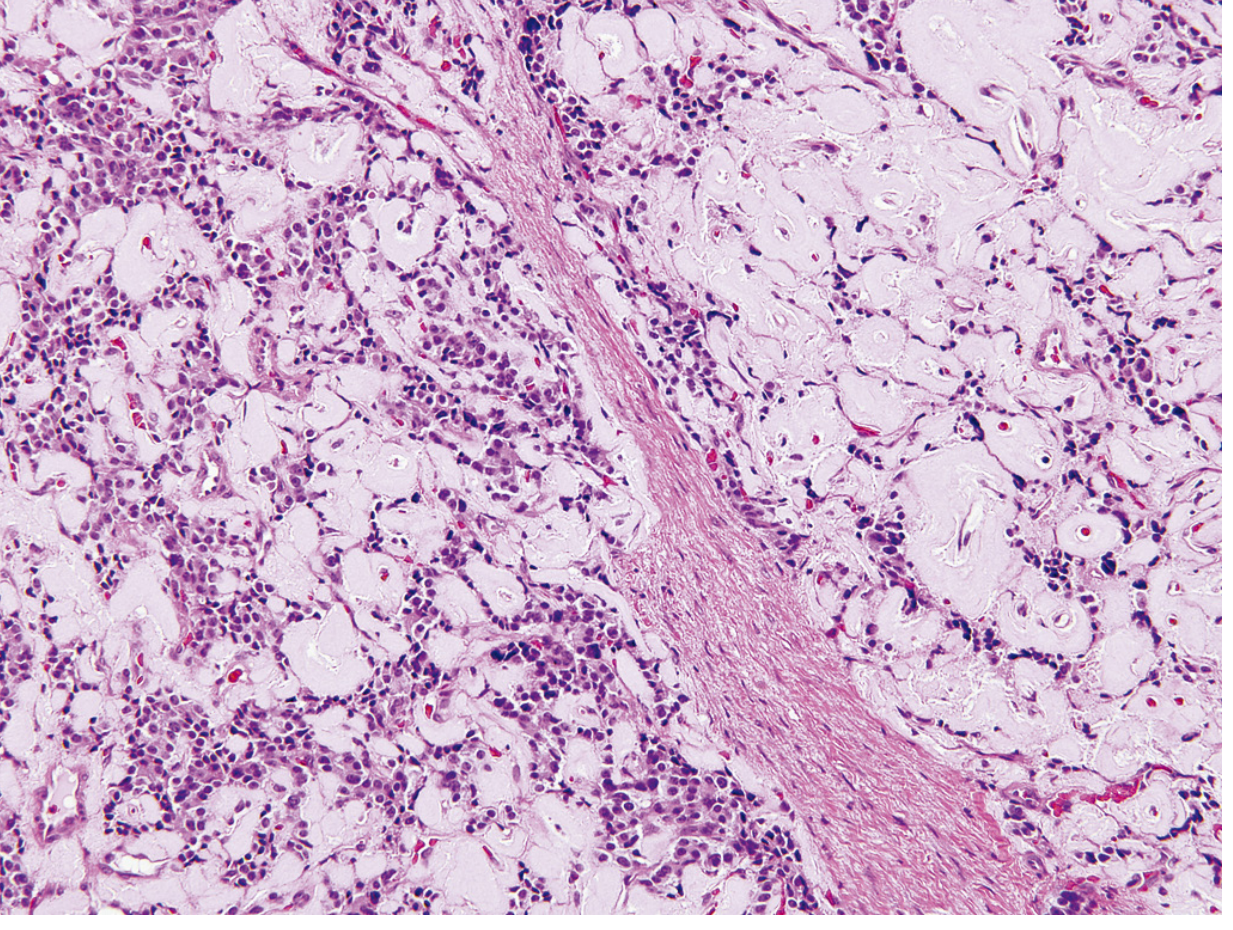
Treatment: Surgical resection - usually followed by prompt reversal of hypoglycemia.
Important differential diagnoses for hypoglycemia: adrenal insufficiency, diffuse liver disease, glycogenoses, ectopic insulin production, sulfonylurea use, factitious hypoglycemia (self-injection of insulin).
2. Zollinger-Ellison Syndrome (Gastrinoma) - p. 1012
Gastrinomas arise from G cells (gastrin-secreting cells) and are the second most common functional PanNEN.
Key Anatomical Point - "Gastrinoma Triangle"
Gastrinomas are equally likely to arise in the duodenum and peripancreatic soft tissues as in the pancreas itself - all within the so-called "gastrinoma triangle."
Pathophysiology
Tumor secretes excess gastrin → stimulates parietal cells → massive gastric acid hypersecretion → severe, refractory peptic ulceration.
Clinical Features
- Peptic ulcers present in 90-95% of patients
- Ulcers are often multiple and occur in unusual locations (e.g., the jejunum, which is rare in ordinary peptic ulcer disease)
- Ulcers are unresponsive to standard therapy - a key clinical clue
- Should be suspected in patients with intractable jejunal ulcers
Morphology
- >50% are locally invasive or metastatic at time of diagnosis
- ~25% arise in the setting of MEN-1 syndrome (multifocal in MEN-1, usually single in sporadic cases)
- Histologically bland, rarely show marked anaplasia
Treatment
- H+/K+-ATPase inhibitors (PPIs) to control acid hypersecretion
- Surgical excision of the neoplasm when possible
- Hepatic metastases carry a poor prognosis - progressive liver failure usually within 10 years
3. Other Rare Pancreatic Endocrine Neoplasms - p. 1012
| Tumor | Cell | Hormone | Clinical Syndrome |
|---|---|---|---|
| Glucagonoma | α cells | Glucagon | Mild diabetes, necrolytic migratory erythema (skin rash), anemia; most common in perimenopausal/postmenopausal women |
| Somatostatinoma | δ cells | Somatostatin | Diabetes mellitus, cholelithiasis, steatorrhea, hypochlorhydria |
| VIPoma | ? | Vasoactive intestinal peptide (VIP) | WDHA syndrome: Watery Diarrhea, Hypokalemia, Achlorhydria; can metastasize |
| Carcinoid | EC cells | Serotonin | Atypical carcinoid syndrome - exceedingly rare in pancreas |
| PPoma | PP cells | Pancreatic polypeptide | Present as mass lesions; even high hormone levels are asymptomatic |
Multihormonal PanNENs also occur - single tumors secreting two or more hormones (e.g., insulin + glucagon + gastrin + ACTH/MSH/ADH/serotonin/norepinephrine). These must be distinguished from the MEN syndromes, where multiple hormones come from tumors in separate glands.
Summary Table
| Feature | Insulinoma | Gastrinoma | Others |
|---|---|---|---|
| Cell of origin | β cell | G cell | α, δ, EC, PP cells |
| Main hormone | Insulin | Gastrin | Glucagon, somatostatin, VIP, etc. |
| Most common subtype? | Yes (#1) | #2 | Rare |
| % Malignant | ~10% (mostly benign) | >50% | High for glucagonoma/VIPoma |
| Key clinical clue | Hypoglycemia relieved by glucose | Refractory/jejunal ulcers | Varies by type |
| MEN-1 association | Yes | Yes (25%) | Yes |
Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 1011-1012
This is a shared conversation. Sign in to Orris to start your own chat.