Congenital adrenal hyperplasia
Reading File
Finding Sources
Searching PubMed
"congenital adrenal hyperplasia"[MeSH Terms]
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed for a comprehensive answer. Let me compose the full response.
Congenital Adrenal Hyperplasia (CAH)
Definition and Pathophysiology
CAH is a group of autosomal recessive disorders caused by defects in the enzymes required for cortisol biosynthesis. The fundamental defect is inadequate cortisol synthesis, which removes the normal negative feedback on the hypothalamic-pituitary axis, leading to elevated CRH and ACTH. The resulting ACTH excess drives adrenal hyperplasia and causes steroid precursors to accumulate proximal to the enzymatic block - many of which are shunted into androgen synthesis.
Adrenal and gonadal steroidogenesis pathway:
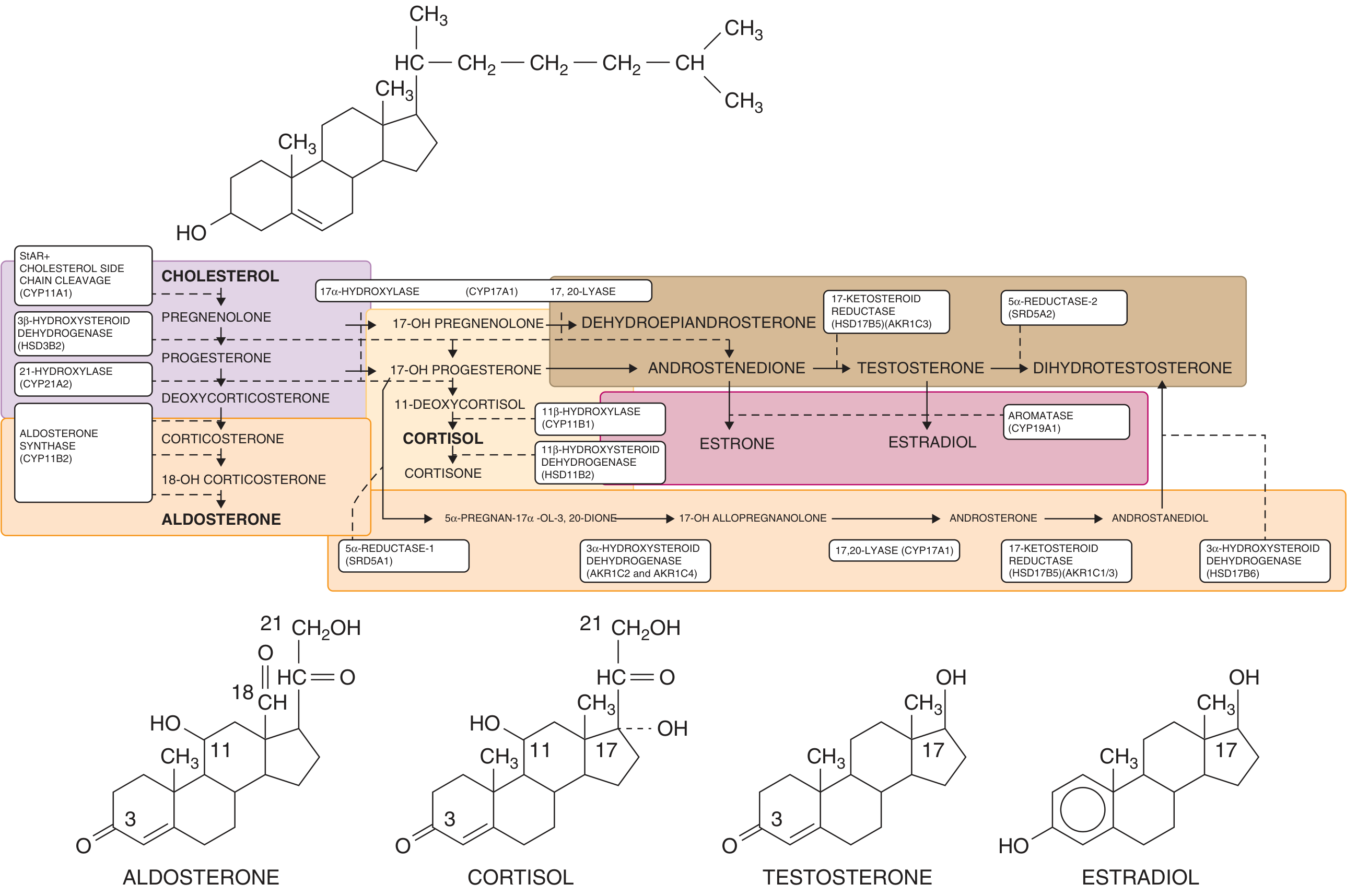
Figure: Adrenal steroidogenesis. Enzymes causing CAH are shown (CYP21A2, CYP11B1, HSD3B2, CYP17A1, CYP11A1). The "backdoor" pathway from 17-OH progesterone to DHT is also shown (orange). - Goldman-Cecil Medicine
Five enzymes are involved in the cortisol biosynthetic pathway. A defect in any of them can cause CAH:
- Cholesterol side-chain cleavage enzyme (CYP11A1 / StAR)
- 3β-hydroxysteroid dehydrogenase (HSD3B2)
- 17α-hydroxylase / 17,20-lyase (CYP17A1)
- 21-hydroxylase (CYP21A2) - the most common
- 11β-hydroxylase (CYP11B1) - second most common
Enzyme Deficiencies
1. 21-Hydroxylase Deficiency (95% of all CAH)
Gene: CYP21A2 on chromosome 6p21.3, within the MHC complex. A highly homologous pseudogene (CYP21A1P, 98% identical) lies adjacent to it; >90% of mutations arise from intergenic recombination between these two genes. Inherited in an autosomal recessive pattern.
Incidence: 1 in 5,000-15,000 in the US/Europe; highest known incidence of 1 in 490 in Yupik Alaskan Eskimos.
Biochemistry: Block at 21-hydroxylation causes accumulation of:
- 17-hydroxyprogesterone (17-OHP) - the key diagnostic marker
- 17-hydroxypregnenolone, progesterone
- These are shunted to DHEA → androstenedione → testosterone (the active androgen in CAH)
- Also via the "backdoor" pathway: progesterone/17-OHP → DHT
Clinical forms:
| Form | % | Aldosterone | Features |
|---|---|---|---|
| Classic - Salt Wasting | ~75% of classic | Deficient | Virilization + life-threatening salt-wasting crises |
| Classic - Simple Virilizing | ~25% of classic | Sufficient | Virilization without salt-wasting |
| Non-Classic (late-onset) | 1/200-1/500 | Normal | Postnatal androgen excess, no prenatal virilization |
Clinical features in 46,XX females (classic):
- Prenatal androgen exposure begins at ~10 weeks' gestation (during external genitalia formation)
- Results in clitoromegaly, labial fusion, urogenital sinus - i.e., ambiguous genitalia (see Prader classification I-V)
- Internal Mullerian structures (uterus, fallopian tubes, ovaries) are normal
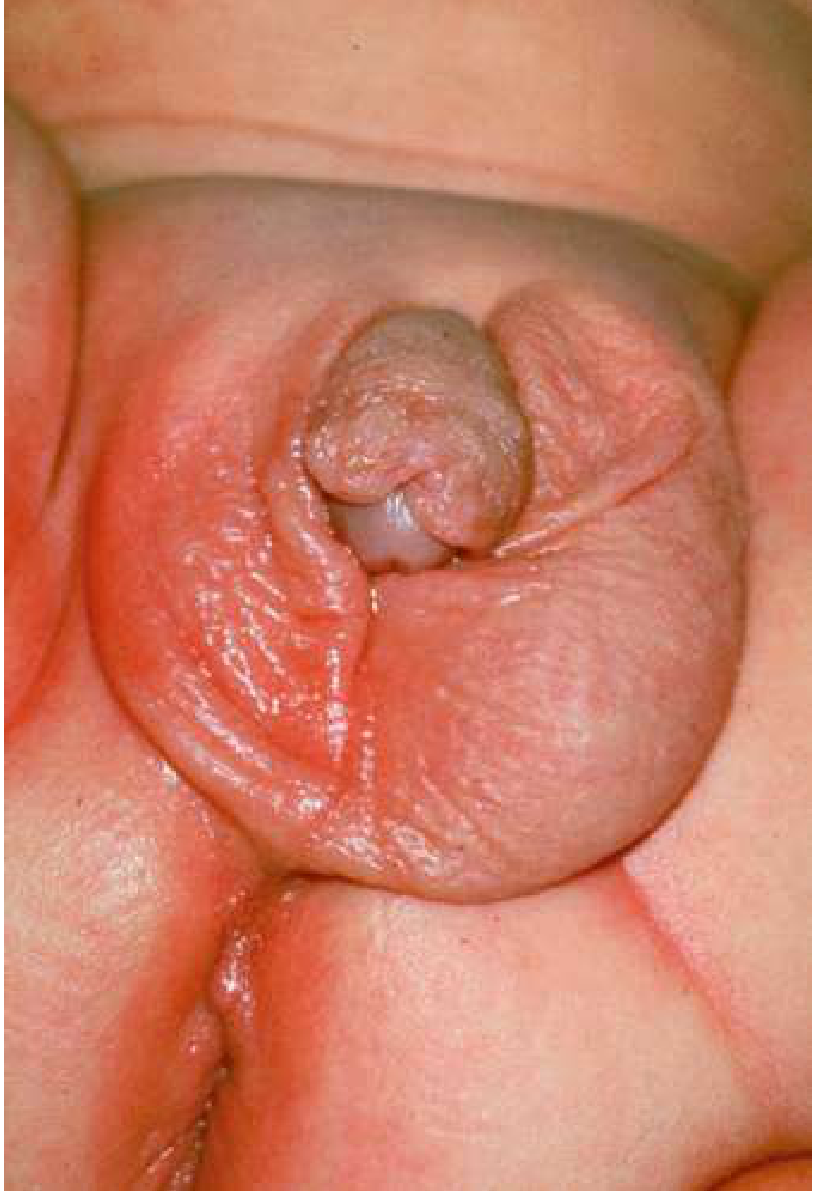
Clinical features in 46,XY males (classic):
- Appear normal at birth (external genitalia are unaffected by excess androgens)
- Salt wasters develop adrenal crisis within 10-21 days of life: vomiting, dehydration, hyperkalemia, shock - may mimic pyloric stenosis
- Simple virilizers: penile and scrotal enlargement, pubic hair, acne, voice deepening, accelerated bone age within first 2-3 years ("little Hercules" musculature)
- Long-term: short stature (premature epiphyseal closure), infertility in 20-40%
- Testicular adrenal rest tumors (TARTs) in up to 30% - diagnosed by scrotal ultrasound
Non-Classic CAH: Presents with signs of androgen excess - premature pubarche, acne, hirsutism, oligomenorrhea, or infertility in women. Many are asymptomatic.
2. 11β-Hydroxylase Deficiency (~5-8% of CAH)
Gene: CYP11B1
Biochemistry: Accumulation of 11-deoxycortisol and deoxycorticosterone (DOC); excess adrenal androgens
Key distinguishing feature from 21-OHD:
- Aldosterone synthesis is unaffected - NO salt wasting
- DOC has mineralocorticoid activity → hypertension (characteristic)
- Virilization is similar to 21-OHD
Diagnosis: Elevated 11-deoxycortisol and 11-deoxycorticosterone (vs. elevated 17-OHP in 21-OHD)
3. Other Rarer Forms
| Deficiency | Key Features |
|---|---|
| 3β-HSD (HSD3B2) | Affects both adrenals and gonads; males under-virilized (some DHT via backdoor), females mildly virilized; salt-wasting; elevated DHEA |
| 17α-Hydroxylase / 17,20-lyase (CYP17A1) | Absent sex hormone synthesis in both sexes; XY = female phenotype; XX = primary amenorrhea; hypertension + hypokalemia (elevated DOC, corticosterone) |
| Lipoid adrenal hyperplasia (StAR mutation) | Most severe; near-total block; both sexes have adrenal insufficiency; XY = female external genitalia; adrenals lipid-laden on imaging |
Prader Classification of Virilization
Prader (1958) classified the degree of virilization of external genitalia in 46,XX females with CAH on a scale of I-V:
- Prader I: Clitoromegaly only, separate vaginal and urethral openings
- Prader II: Clitoromegaly, single perineal opening (urogenital sinus)
- Prader III: Enlarged phallus with urogenital sinus at base
- Prader IV: Penile urethra opens near the base of a male-appearing phallus
- Prader V: Complete male-appearing external genitalia (phallus with penile urethra at tip, empty "scrotum")
The degree of genital ambiguity correlates with the severity of the underlying mutation.
Diagnosis
Neonatal Screening
- Elevated 17-OHP on blood spot (filter paper) - standard of care in most countries
- Premature and sick infants may have false-positive elevations
Biochemical Evaluation
- Morning plasma 17-OHP: markedly elevated in classic 21-OHD (often >10,000 ng/dL)
- For non-classic CAH: basal 17-OHP <200 ng/dL (6 nmol/L) essentially rules out non-classic 21-OHD; if borderline, do ACTH stimulation test (250 mcg cosyntropin) with 1-hour 17-OHP
- Urinary 17-ketosteroids and pregnanetriol: elevated in classic disease
- Plasma renin activity: elevated in salt-wasting form (reflects aldosterone deficiency)
- Electrolytes: hyponatremia, hyperkalemia in salt-wasters
- Serum androstenedione, DHEA, testosterone: elevated
For 11β-OHD: elevated 11-deoxycortisol and 11-deoxycorticosterone; elevated 17-OHP but less dramatically than 21-OHD
Imaging and Other Workup
- Karyotype: mandatory in any infant with ambiguous genitalia
- Pelvic ultrasound: confirm presence of uterus/ovaries in virilized 46,XX infant
- Scrotal ultrasound: screen for TARTs in males
- Bone age (wrist X-ray): assess degree of advancement from androgen excess
Management
Acute Adrenal Crisis (Salt-Wasting)
- IV hydrocortisone (stress dosing: 50-100 mg/m²/day or 1-2 mg/kg bolus)
- IV normal saline and dextrose for hyponatremic dehydration
- Correct hyperkalemia
Long-Term Medical Therapy
Glucocorticoid replacement - primary treatment for all forms:
- Hydrocortisone is preferred in children (least effect on growth suppression)
- Typical dose: 10-15 mg/m²/day in 2-3 divided doses
- Prednisolone or dexamethasone in adults (longer acting, better suppression of ACTH/androgens)
- Goal: suppress excess adrenal androgens while avoiding over-suppression (Cushingoid features, growth suppression)
- Monitoring: 17-OHP, androstenedione, bone age, growth velocity
Mineralocorticoid replacement (salt-wasting form):
- Fludrocortisone (9α-fluorohydrocortisone): 0.05-0.2 mg/day
- Sodium supplementation in infants
- Monitor plasma renin activity, blood pressure
Non-classic CAH: Treatment only if symptomatic (hirsutism, menstrual irregularities, infertility); low-dose glucocorticoids or other anti-androgen therapy
Stress dosing: Patients must receive 2-3× usual glucocorticoid dose during illness, injury, or surgery ("sick day rules") to prevent adrenal crisis
Surgical Management (in virilized 46,XX females)
- Feminizing genitoplasty: clitoral reduction and vaginoplasty if indicated
- Timing and extent are individualized; multidisciplinary approach with endocrinology, urology, psychology
- Surgical intervention is typically deferred, particularly for vaginoplasty, until later childhood or adolescence when possible
Prenatal Treatment (experimental/controversial)
- Dexamethasone given to the mother starting before 9 weeks of gestation (before sex can be determined)
- Crosses the placenta, suppresses fetal ACTH, reduces virilization in 46,XX fetuses
- Given to 7 out of 8 fetuses unnecessarily (since only 1 in 8 pregnancies produces an affected female)
- Now not routinely recommended outside of research protocols due to concerns about maternal and fetal adverse effects
Psychological and Behavioral Aspects
CAH in 46,XX individuals is associated with:
- Gender role: preference for masculine-typical toys/activities ("tomboyish" behavior), less interest in child rearing
- Sexual orientation: higher rates of bisexual/homosexual orientation in women with salt-wasting form (proposed to reflect prenatal androgen imprinting of the brain)
- Gender identity: 4% of 46,XX CAH women report gender dysphoria; higher rates (15%) when raised as males
- Psychosocial support is an integral part of long-term management
Key Points Summary
| Feature | 21-OHD | 11β-OHD |
|---|---|---|
| Frequency | 95% | ~5% |
| Gene | CYP21A2 (chr 6p21.3) | CYP11B1 |
| Key marker | ↑17-OHP | ↑11-deoxycortisol, ↑DOC |
| Salt-wasting | Yes (75% of classic) | No |
| BP | Normal/low | Hypertension |
| Virilization | Yes | Yes |
| Treatment | Hydrocortisone ± fludrocortisone | Hydrocortisone |
- Goldman-Cecil Medicine, pp. 2515-2522
- Campbell-Walsh-Wein Urology, pp. 1291-1295
Addison disease
Finding Sources
Searching PubMed
"Addison disease"[MeSH Terms] AND "adrenal insufficiency"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive information from multiple authoritative sources. Let me compose the full answer.
Addison Disease (Primary Adrenal Insufficiency)
Definition
Addison disease is primary adrenocortical insufficiency - failure of the adrenal cortex itself to produce adequate amounts of cortisol and aldosterone despite normal or elevated ACTH stimulation. It is distinct from secondary adrenal insufficiency (pituitary/hypothalamic failure). Clinically evident disease does not appear until ≥90% of adrenal cortical tissue has been destroyed.
Classification
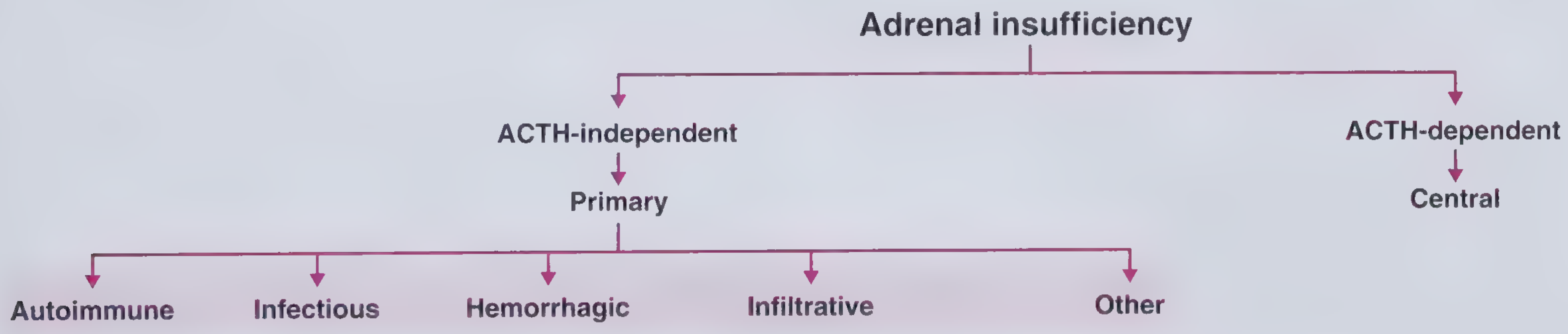
Figure: Classification of adrenal insufficiency. - Frameworks for Internal Medicine
Normal HPA Axis (for reference)
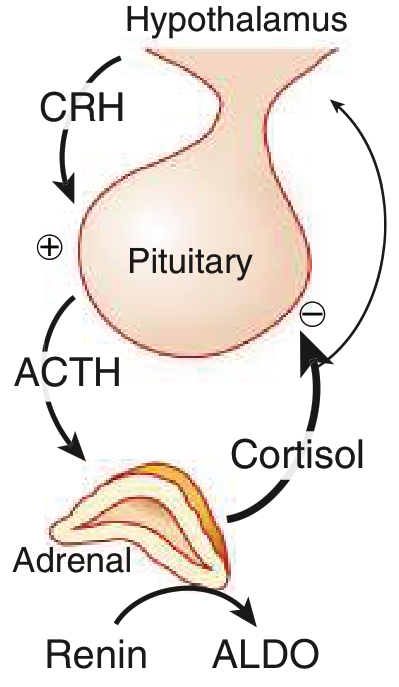
In primary adrenal insufficiency, the adrenal gland fails, cortisol and aldosterone fall, negative feedback is lost, and ACTH rises. Mineralocorticoid secretion is largely independent of ACTH (controlled by the renin-angiotensin system), so in secondary insufficiency, aldosterone is typically preserved.
Etiology
1. Autoimmune (most common in industrialized world, ~80%)
- Isolated autoimmune adrenalitis - most common
- Polyglandular Autoimmune Syndrome Type 1 (PAS-1 / APS-1 / APECED):
- Onset <20 years
- Triad: hypoparathyroidism + mucocutaneous candidiasis + adrenal insufficiency
- Due to AIRE gene mutation
- Polyglandular Autoimmune Syndrome Type 2 (PAS-2):
- Onset >40 years
- Associated with insulin-dependent diabetes mellitus, autoimmune thyroid disease (Hashimoto/Graves), alopecia areata, vitiligo
- Marker: anti-21-hydroxylase antibodies (present in nearly all autoimmune cases)
- Imaging: small adrenal glands
2. Infectious (~15% in US series)
- Tuberculosis - historically most common worldwide; adrenal tissue replaced by caseating granulomas; may show calcification on imaging
- Systemic fungal infections: histoplasmosis, paracoccidioidomycosis, cryptococcosis, blastomycosis, coccidioidomycosis
- AIDS-associated: CMV infiltration, Kaposi sarcoma, lymphoma, or drug effects (ketoconazole, rifampin)
- Early/recent infection (<2 years): bilateral adrenal enlargement; long-standing: calcification and atrophy
3. Hemorrhagic
- The adrenal gland is susceptible to hemorrhage because 3 arteries supply it but only 1 vein drains it - a "vascular dam" that predisposes to hemorrhage during hemodynamic stress
- Risk factors: thromboembolic disease, coagulopathy, anticoagulation (e.g., heparin), postoperative state, sepsis
- Waterhouse-Friderichsen syndrome: bilateral adrenal hemorrhage in the setting of fulminant meningococcemia
- Diagnosis: biochemical adrenal insufficiency + CT/MRI/ultrasound evidence of hemorrhage
- Prognosis: high mortality if missed
4. Infiltrative
- Bilateral metastases (lung, breast, kidney, gut carcinoma; lymphoma) - adrenal insufficiency uncommon unless extensive
- Amyloidosis
- Sarcoidosis
5. Other Causes
- Adrenoleukodystrophy (ALD): X-linked, 1 in 25,000; deficiency of peroxisomal membrane protein (ABCD1 gene) → accumulation of very long chain fatty acids (VLCFA) → inhibit ACTH signal transduction in adrenal cortex; also causes CNS demyelination (progressive spastic paraparesis, cognitive decline). In boys with Addison disease, must always be excluded. Elevated plasma C26:0 fatty acids is the diagnostic test.
- Drugs: Ketoconazole, mitotane, aminoglutethimide, trilostane, metyrapone (steroidogenesis inhibitors); etomidate (single dose can suppress cortisol)
- Congenital adrenal hypoplasia
- ACTH resistance (familial glucocorticoid deficiency)
Secondary Adrenal Insufficiency (not Addison, for comparison)
- Most common cause: exogenous glucocorticoid use (HPA axis suppression; typically at doses >15-20 mg hydrocortisone equivalent for >3 weeks)
- Structural pituitary/hypothalamic lesions: tumors, trauma, radiotherapy, lymphocytic hypophysitis, infiltration
- Key difference from Addison disease: no hyperpigmentation; no mineralocorticoid deficiency (aldosterone preserved)
Pathophysiology and Clinical Features
Loss of all three adrenocortical hormone classes produces the characteristic syndrome:
| Hormone lost | Consequences |
|---|---|
| Cortisol (glucocorticoid) | Fatigue, weakness, hypoglycemia, anorexia, weight loss, nausea, vomiting, loss of stress response |
| Aldosterone (mineralocorticoid) | Hyponatremia, hyperkalemia, metabolic acidosis, ECF volume depletion, hypotension, orthostatic hypotension, salt craving |
| DHEA/androstenedione (adrenal androgens) | Decreased pubic and axillary hair, decreased libido (in women, since these are the primary androgen source) |
| ACTH excess (loss of negative feedback) | Hyperpigmentation - via MSH activity of ACTH/POMC |
Hyperpigmentation - a Hallmark of Primary Adrenal Insufficiency
ACTH is derived from the POMC precursor, which also contains α-MSH (melanocyte-stimulating hormone). Elevated ACTH → elevated MSH → increased melanocyte stimulation.
Hyperpigmentation is:
- Diffuse, but most pronounced in:
- Sun-exposed areas
- Pressure/friction sites: elbows, knees, knuckles
- Skin creases (especially palmar creases)
- Recent scars (a particularly useful sign)
- Mucous membranes (buccal mucosa, gingiva, vaginal mucosa) - a key diagnostic pointer
- Nipples and areolae
- Nail beds
- Hyperpigmentation is NOT present in secondary adrenal insufficiency (ACTH is low or normal in those cases) - this distinction is diagnostically critical
Acute Adrenal Crisis (Addisonian Crisis)
- May be the first presentation, or may be precipitated by intercurrent illness/surgery in a known patient
- Features: orthostatic hypotension → cardiovascular collapse, abdominal pain, agitation, confusion, fever
- Labs: severe hyponatremia, hyperkalemia, hypoglycemia, eosinophilia, prerenal azotemia
- Life-threatening emergency - must treat immediately without waiting for confirmatory tests
Chronic Presentation
- Gradual onset: malaise, fatigue, weight loss, anorexia, arthralgias, back pain, GI symptoms
- Salt craving (sometimes extreme - e.g., craving pickle brine)
- Hyperpigmentation
- Vitiligo may be associated (in autoimmune cases)
- Associated features of polyglandular syndrome (hypothyroidism, diabetes, hypoparathyroidism)
Biochemical Features
| Finding | Explanation |
|---|---|
| ↓ Serum sodium (hyponatremia) | Aldosterone deficiency → sodium loss; also cortisol deficiency → inappropriate ADH |
| ↑ Serum potassium (hyperkalemia) | Aldosterone deficiency → K+ retention |
| Hypoglycemia | Cortisol deficiency → reduced gluconeogenesis |
| Metabolic acidosis | Aldosterone deficiency → H+ retention |
| Eosinophilia | Cortisol normally suppresses eosinophils; cortisol deficiency → eosinophilia |
| Mild prerenal azotemia | Volume depletion |
| ↑ ACTH | Loss of cortisol negative feedback on pituitary |
| ↓ Aldosterone + ↑ Renin | Primary adrenal failure affecting mineralocorticoid zone |
Diagnosis
Screening Test
- Morning serum cortisol:
-
18 μg/dL (500 nmol/L): virtually excludes adrenal insufficiency
- <3 μg/dL (83 nmol/L): strongly suggests adrenal insufficiency
- 3-18 μg/dL: indeterminate → needs stimulation testing
-
Gold Standard: ACTH (Cosyntropin) Stimulation Test
- Give 250 μg cosyntropin (synthetic ACTH 1-24) IV at any time of day
- Measure serum cortisol at 30 and 60 minutes
- Normal response: cortisol >18 μg/dL (by immunoassay); or >14 μg/dL by mass spectrometry/monoclonal assay
- Failure to reach this threshold = adrenal insufficiency
- A 1-μg dose may be used in the morning and is equally diagnostic, but dose preparation is more error-prone
Metyrapone Test
- Useful for mild or recent secondary adrenal insufficiency (where ACTH response may be preserved)
- Weight-based dose at midnight; cortisol and 11-deoxycortisol measured the next morning
- 11-deoxycortisol <7 μg/dL = adrenal insufficiency
Distinguishing Primary from Secondary
- Plasma ACTH: markedly elevated (>2× upper normal) in primary; low/normal in secondary
- Aldosterone + plasma renin activity: low aldosterone with high renin = primary (mineralocorticoid zone affected); normal in secondary
- Imaging:
- Primary autoimmune: small adrenal glands
- Primary infectious/infiltrative/hemorrhagic: large or calcified adrenals
- Secondary: normal or small adrenals; pituitary/hypothalamic lesion on MRI
Identifying the Cause
- Anti-21-hydroxylase antibodies: present in nearly all autoimmune primary adrenal insufficiency
- If antibody-negative in a male: plasma VLCFA (C26:0) to exclude adrenoleukodystrophy
- CT adrenal imaging: to exclude infection, metastases, hemorrhage
- Screen for other autoimmune endocrinopathies (thyroid, parathyroid, glucose)
Treatment
Acute Adrenal Crisis - Emergency Management
- Do not delay treatment - draw blood for cortisol measurement, then treat immediately
- IV Hydrocortisone 100 mg bolus (has both glucocorticoid and mineralocorticoid activity at this dose)
- IV normal saline (1-2 L rapidly) for volume expansion
- IV dextrose (D5NS) for hypoglycemia
- Continue hydrocortisone 50-100 mg IV every 6-8 hours or as continuous infusion
- Identify and treat the precipitating cause (infection, etc.)
- Transition to oral replacement once stable
Chronic Glucocorticoid Replacement
- Hydrocortisone (preferred): 10-12 mg/m²/day in 2-3 divided doses
- Largest dose in the morning (mimics the normal diurnal cortisol peak)
- One-third of daily dose given around 4 PM if split-dosing
- Taking the morning dose 30 min before rising helps those with severe morning fatigue
- Prednisolone 4-7 mg daily: longer half-life, useful for afternoon/evening fatigue
- Dexamethasone: infrequently used (dose difficult to standardize)
- Monitoring: clinical assessment, electrolytes, bone density; avoid over-replacement (Cushingoid side effects)
Mineralocorticoid Replacement (primary adrenal insufficiency)
- Fludrocortisone (9α-fluorohydrocortisone) 50-300 μg/day orally
- Dose titrated to: normal blood pressure, normal serum sodium and potassium, normal plasma renin activity
- Patients should be encouraged to eat salt liberally and not restrict dietary sodium
- Note: Mineralocorticoid replacement is NOT needed in secondary adrenal insufficiency
DHEA Replacement
- Some patients with primary adrenal insufficiency have low DHEA
- Routine replacement is not supported by current evidence
Sick Day Rules and Stress Dosing - Critical Patient Education
- All patients must know to double or triple their glucocorticoid dose during:
- Febrile illness (>38°C)
- Vomiting/diarrhea (may need IM/IV hydrocortisone if unable to take oral medication)
- Significant physical stress, trauma, surgery
- Patients should carry a steroid alert card and/or a medical alert bracelet
- IM hydrocortisone for self-injection should be prescribed for emergencies
Primary vs Secondary Adrenal Insufficiency - Key Differences
| Feature | Primary (Addison) | Secondary |
|---|---|---|
| Site of defect | Adrenal cortex | Pituitary / Hypothalamus |
| ACTH | ↑↑ (elevated) | ↓ or normal |
| Cortisol | ↓ | ↓ |
| Aldosterone | ↓ (affected) | Normal |
| Plasma renin | ↑ | Normal |
| Hyperpigmentation | YES | No |
| Electrolytes | Hyponatremia, hyperkalemia | Hyponatremia only (mild) |
| Most common cause | Autoimmune destruction | Exogenous glucocorticoids |
| Mineralocorticoid Rx needed | Yes | No |
Recent Evidence
Two recent systematic reviews (2024-2025) are relevant:
- Ilia et al., 2024 (PMID 39277840) found increased prevalence of adverse pregnancy and fetal outcomes in women with primary adrenal insufficiency - counseling on sick-day rules and stress dosing during pregnancy is particularly important.
- Dalakas et al., 2025 (PMID 40966723) confirmed increased all-cause mortality in primary adrenal insufficiency - likely driven by adrenal crises, cardiovascular events, and infections - reinforcing the need for optimized replacement therapy and crisis prevention.
Sources: Goldman-Cecil Medicine, pp. 2456-2457 | Costanzo Physiology, 7th Ed., p. 440 | Frameworks for Internal Medicine, pp. 98-101 | Andrews' Diseases of the Skin, p. 579
This is a shared conversation. Sign in to Orris to start your own chat.