Teach me everything I need to know as an MBBS student on Single Gene Genetic Disorders (Robbins pathology)
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed for a comprehensive, well-structured answer from Robbins Pathology. Let me compile it.
Single-Gene (Mendelian) Genetic Disorders
Robbins & Kumar Basic Pathology — Complete MBBS Guide
1. OVERVIEW & KEY CONCEPTS
Single-gene mutations follow Mendelian inheritance and are individually rare but collectively impose a significant disease burden. The OMIM database catalogs these disorders.
Critical tenets:
- Pleiotropy: One mutation → multiple phenotypic effects (e.g., Marfan syndrome: skeleton + eyes + cardiovascular from a single FBN1 mutation)
- Genetic heterogeneity: Mutations at different loci → same phenotype (e.g., retinitis pigmentosa)
- Modifier genes: Other loci influence severity (important in cystic fibrosis)
- Prenatal screening in high-risk populations (e.g., Ashkenazi Jewish) has reduced incidence of disorders like Tay-Sachs
2. PREVALENCE TABLE (Selected Disorders)
| Disorder | Inheritance | Estimated Prevalence |
|---|---|---|
| Familial hypercholesterolemia | AD | 1 in 500 |
| Polycystic kidney disease | AD | 1 in 1,000 |
| Marfan syndrome | AD | 1 in 5,000 |
| Huntington disease | AD | 1 in 10,000 |
| Sickle cell anemia | AR | 1 in 500 (US, African descent) |
| Cystic fibrosis | AR | 1 in 3,200 (N. European descent) |
| Tay-Sachs disease | AR | 1 in 3,500 (Ashkenazi Jewish) |
| Phenylketonuria | AR | 1 in 10,000 |
| Duchenne muscular dystrophy | X-linked | 1 in 3,500 (males) |
3. TRANSMISSION PATTERNS
A. Autosomal Dominant (AD)
- Manifest in heterozygous state
- Both sexes affected and can transmit
- 50% offspring of an affected person are affected
- Key features:
- New mutations possible → affected child with unaffected parents (siblings unaffected)
- Reduced penetrance: carries mutant gene but phenotypically normal
- Variable expressivity: same gene, different severity (e.g., neurofibromatosis 1 — from skin spots to multiple tumors)
- Delayed onset: symptoms may not appear until adulthood (e.g., Huntington, adult-onset cancer genes)
- Genes involved are usually not enzymes but: regulatory/signaling proteins, structural proteins, receptors/transport proteins (because 50% loss of enzyme activity is compensable)
B. Autosomal Recessive (AR)
- Manifest in homozygous state
- Often involves enzyme deficiencies
- Parents are typically unaffected carriers
- 25% offspring of two carriers are affected
- Often more severe than dominant disorders
- Consanguinity increases risk
C. X-Linked Recessive
- Heterozygous female carriers transmit to sons (50% sons affected)
- Males are hemizygous → fully express the disorder
- Affected male does NOT transmit to sons; all daughters are carriers
- Females rarely express full phenotype (random X inactivation protects most cells)
- Examples: Duchenne muscular dystrophy, hemophilia A & B, G6PD deficiency
Biochemical basis table:
| Disease | Abnormal Protein | Protein Type | Inheritance |
|---|---|---|---|
| Familial hypercholesterolemia | LDL receptor | Receptor/transport | AD |
| Marfan syndrome | Fibrillin | ECM structural | AD |
| Ehlers-Danlos syndrome | Collagen | ECM structural | AD/AR |
| Cystic fibrosis | CFTR | Ion channel | AR |
| Phenylketonuria | Phenylalanine hydroxylase | Enzyme | AR |
| Tay-Sachs | Hexosaminidase | Enzyme | AR |
4. DISEASES CAUSED BY MUTATIONS IN STRUCTURAL PROTEIN GENES
4A. Marfan Syndrome
- Inheritance: Autosomal dominant
- Gene: FBN1 at 15q21 (encodes fibrillin-1, an extracellular glycoprotein that is the major component of microfibrils)
- Prevalence: 1 in 5,000; 70–85% familial, rest sporadic de novo mutations
- Pathogenesis:
- Microfibrils serve as scaffolds for tropoelastin (elastic fibers)
- Mutant fibrillin acts as dominant negative → impairs microfibril assembly
- Loss of microfibrils → excessive TGF-β activation (microfibrils normally sequester TGF-β) → vascular smooth muscle damage and ECM degradation
- Mutations in TGF-β type II receptor → Marfan syndrome type 2 (MFS2)
Morphology (MBBS exam key features):
| System | Features |
|---|---|
| Skeleton | Tall stature; arachnodactyly (long fingers/toes); dolichostenomelia (long limbs); high-arched palate; kyphoscoliosis; pectus excavatum/carinatum |
| Eyes | Bilateral ectopia lentis (upward lens dislocation) in 50–80% |
| Cardiovascular | Mitral valve prolapse (most common lesion); aortic regurgitation; cystic medial degeneration of aorta → aortic root dilation → aortic dissection (leading cause of death) |
Treatment: β-blockers + angiotensin receptor blockers (both inhibit TGF-β signaling) to reduce cardiovascular risk.
4B. Ehlers-Danlos Syndromes (EDS)
A clinically and genetically heterogeneous group of disorders caused by defects in collagen synthesis or structure.
| EDS Type | Defect | Inheritance | Key Feature |
|---|---|---|---|
| Vascular EDS | Mutation in COL3A1 (type III collagen) | AD | Blood vessel/bowel rupture |
| Kyphoscoliotic EDS | Lysyl hydroxylase deficiency → impaired collagen crosslinks | AR | Congenital scoliosis, ocular fragility |
| Classical EDS | Mutations in COL5A1/COL5A2 (type V collagen) | AD | Hyperextensible skin, hypermobile joints |
General features: Hyperextensible skin, hypermobile joints, poor wound healing with tissue paper-like scars, bleeding tendency.
Risk: Vascular EDS has rupture risk in surgery; wound healing complications are universal.
5. DISEASES CAUSED BY MUTATIONS IN RECEPTOR/CHANNEL GENES
5A. Familial Hypercholesterolemia (FH)
- Inheritance: Autosomal dominant
- Gene: Loss-of-function mutations in LDL receptor gene (80–85% of cases)
- Prevalence: Heterozygous 1 in 500 (most common Mendelian disorder); prevalence in cardiovascular disease is 20-fold higher
Normal LDL Metabolism:
- Liver secretes VLDL → lipolysis in capillaries → IDL → further metabolism → LDL
- LDL receptor (binds Apo B-100 and Apo E) on hepatocytes (75% of receptors) mediates LDL uptake
- Intracellular cholesterol causes: ↓ HMG-CoA reductase (↓ cholesterol synthesis), ↓ LDL receptor synthesis (feedback), ↑ ACAT (cholesterol esterification for storage)
Pathogenesis of FH:
- Receptor mutation → impaired LDL clearance → elevated plasma LDL
- Loss of feedback inhibition → unchecked cholesterol synthesis
- LDL accumulates in macrophages → foam cells → atherosclerosis
Mutations in LDL receptor — 5 classes:
- Receptor not synthesized
- Failure to transport to Golgi
- Failure to bind LDL
- Failure to cluster in coated pits
- Failure to release LDL in endosomes
Clinical features:
| Feature | Heterozygotes (1 in 500) | Homozygotes (1 in 1 million) |
|---|---|---|
| Plasma LDL | 2–3× normal | 5–6× normal |
| Xanthomas | Tendon xanthomas (adult onset) | Cutaneous + tendon xanthomas in childhood |
| MI risk | By age 40–50 | MI in childhood; before age 20 |
5B. Cystic Fibrosis (CF)
- Inheritance: Autosomal recessive (most common life-shortening AR disorder in white populations)
- Gene: CFTR on chromosome 7q31 (encodes the Cystic Fibrosis Transmembrane Regulator, a cAMP-regulated Cl⁻ channel)
- Most common mutation: ΔF508 (deletion of phenylalanine at position 508) → misfolded protein degraded in ER; accounts for ~70% of cases
- Modifier genes: Genes encoding TGF-β, mannose-binding lectin, and other loci modulate disease severity
Pathogenesis:
- CFTR normally secretes Cl⁻ into airway lumen → draws water → thin, watery mucus
- In CF: Cl⁻ secretion blocked → Na⁺ reabsorption increased → water absorbed from lumen → thick, viscid mucus
- Thick mucus → plugging → obstruction → infection (especially Pseudomonas aeruginosa, Staphylococcus aureus) → chronic inflammation → bronchiectasis
Morphology & Clinical Features:
| Organ | Pathology | Consequence |
|---|---|---|
| Lungs | Mucus plugging, bronchiectasis, recurrent bacterial infections | Most common cause of death; respiratory failure |
| Pancreas | Ductal obstruction → acinar destruction → fibrosis → exocrine pancreatic insufficiency | Malabsorption, steatorrhea |
| Liver | Biliary cirrhosis (focal) | Hepatic failure (minority) |
| Intestine | Meconium ileus in 5–10% neonates | Neonatal intestinal obstruction |
| Vas deferens | Bilateral congenital absence | Infertility in males (98%) |
| Sweat glands | Impaired Cl⁻ reabsorption → ↑ Cl⁻ in sweat | Diagnostic: sweat chloride >60 mEq/L |
CFTR mutations — 6 classes:
- No CFTR produced
- CFTR misfolded → retained in ER (ΔF508)
- CFTR reaches surface but cannot open
- CFTR channel conducts Cl⁻ poorly
- Reduced CFTR production
- CFTR at surface but unstable
Treatment advances: CFTR modulators (e.g., ivacaftor, lumacaftor/ivacaftor) that target Class 3 and Class 2 mutations are now in clinical use.
6. DISEASES CAUSED BY ENZYME MUTATIONS
6A. Phenylketonuria (PKU)
- Inheritance: Autosomal recessive
- Gene: Phenylalanine hydroxylase (PAH)
- Prevalence: 1 in 10,000 (European descent)
Pathogenesis:
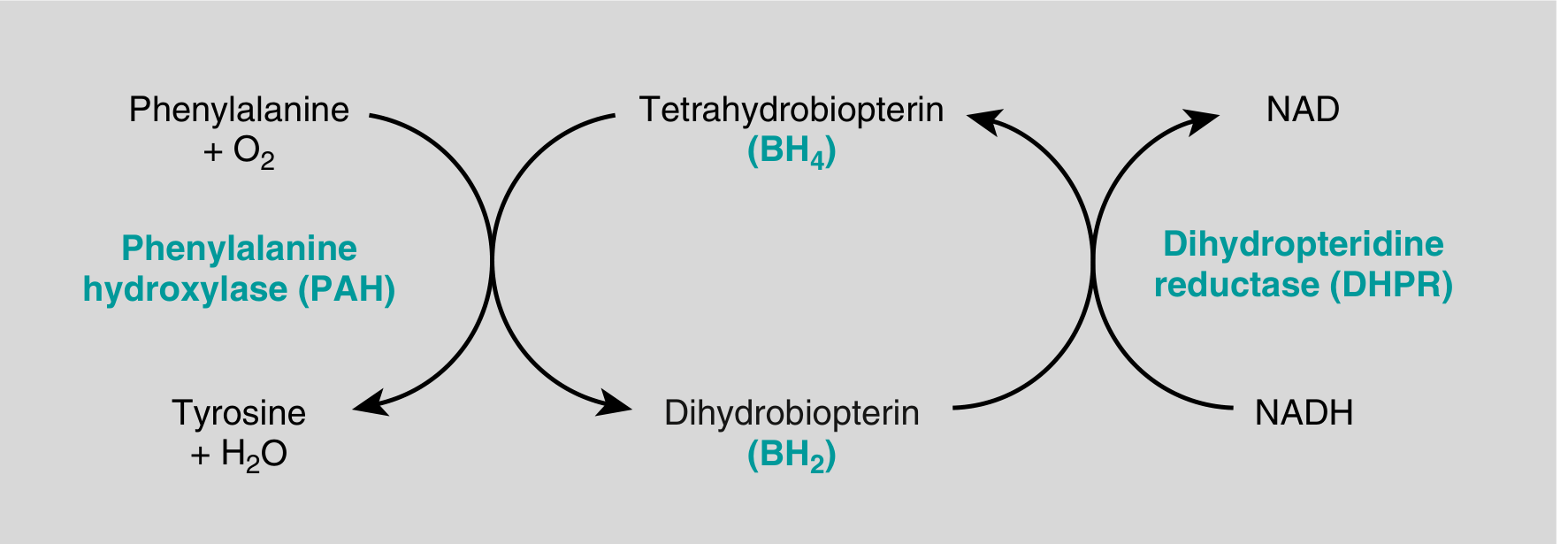
- PAH converts phenylalanine → tyrosine using cofactor BH₄ (tetrahydrobiopterin); DHPR regenerates BH₄
- In PKU: PAH absent → hyperphenylalaninemia → shunt pathways produce phenylpyruvate, phenylacetate, phenyllactate (excreted in urine/sweat → "mousy/musty odor")
- Excess phenylalanine → impairs brain development (mechanism: competitive inhibition of amino acid transport, reduced myelin synthesis)
- ↓ Tyrosine → ↓ melanin → fair skin/hair (even in dark-skinned individuals)
Clinical features (untreated):
- Normal at birth
- Rising plasma phenylalanine within weeks
- Severe intellectual disability (IQ <60) by 6 months
- Seizures, eczema, hypopigmentation
- Musty body odor
Special variant — Maternal PKU: Women with treated PKU who stop dietary restriction in adulthood → phenylalanine crosses placenta → fetal damage → 75–90% of offspring have intellectual disability + microcephaly + congenital heart disease (even though offspring are heterozygotes)
Screening & Treatment:
- Newborn screening: Guthrie test (heel-prick blood spot) — mandatory in most countries
- Treatment: phenylalanine-restricted diet for life
- BH₄ supplementation (sapropterin) works for some mutations
6B. Galactosemia
- Inheritance: AR
- Enzyme defect: Galactose-1-phosphate uridyltransferase (classic form)
- Galactose-1-phosphate accumulates → toxicity in liver, brain, kidney
- Clinical features: Jaundice, hepatomegaly, cataracts, intellectual disability, increased risk of E. coli sepsis in neonates
- Newborn screening + galactose-free diet prevents complications
7. LYSOSOMAL STORAGE DISEASES
All are autosomal recessive. Result from deficiency of lysosomal enzymes → accumulation of undigested metabolites in lysosomes → organ/tissue damage.
General Mechanism:
Enzyme deficiency → substrate accumulates in lysosomes → cell dysfunction → organomegaly (liver, spleen, lymph nodes, bone marrow) and/or CNS damage
Key Diseases:
7A. Tay-Sachs Disease (GM2 Gangliosidosis)
- Deficient enzyme: Hexosaminidase A (HexA) — α-subunit encoded by HEXA gene
- Accumulated substrate: GM2 ganglioside (in neurons)
- Prevalence: 1 in 3,500 (Ashkenazi Jewish)
- Features:
- Normal at birth; progressive neurodegeneration from ~6 months
- Hypotonia → dementia → blindness → paralysis → death by age 2–3
- Cherry-red spot on macula (ganglion cells of retina replaced by lipid-laden neurons; fovea, which lacks these cells, appears cherry-red against pale surroundings)
- No organomegaly (unlike Niemann-Pick)
- Diagnosis: HexA activity in leukocytes; carrier screening in Ashkenazi Jews (reduced Tay-Sachs incidence by prenatal screening programs)
7B. Niemann-Pick Disease
Three distinct types:
| Type | Defect | Accumulated substrate | Key Features |
|---|---|---|---|
| Type A | Sphingomyelinase deficiency | Sphingomyelin + cholesterol | Severe infantile neurodegeneration, hepatosplenomegaly, cherry-red spot |
| Type B | Sphingomyelinase deficiency (partial) | Sphingomyelin | Organomegaly, no CNS involvement |
| Type C | NPC1/NPC2 mutations → defective intracellular cholesterol transport | Cholesterol + gangliosides (GM1/GM2) | Ataxia, vertical supranuclear gaze palsy, dystonia, psychomotor regression |
Diagnosis: sphingomyelinase activity in leukocytes; molecular genetic testing
7C. Gaucher Disease (Most common lysosomal storage disease)
- Deficient enzyme: Glucocerebrosidase (glucocerebrosidase, encoded by GBA gene)
- Accumulated substrate: Glucocerebroside in mononuclear phagocytes (macrophages in liver, spleen, bone marrow)
- Gaucher cells: Enlarged macrophages with "wrinkled tissue paper" cytoplasm (distended lysosomes)
- Pathology is dual: Storage burden + macrophage activation (high IL-1, IL-6, TNF)
| Type | Features |
|---|---|
| Type 1 (99% of cases, chronic non-neuronopathic) | Hepatosplenomegaly (massive spleen), bone involvement (osteopenia, osteonecrosis), cytopenias, no CNS involvement; compatible with long life; carrier frequency 1:12 in Ashkenazi Jews |
| Type 2 (acute neuronopathic) | Infancy onset; severe CNS involvement; death before age 2 |
| Type 3 (subacute neuronopathic) | Onset childhood/adolescence; milder CNS signs |
Treatment: Enzyme replacement therapy (imiglucerase/velaglucerase) effective for Type 1.
7D. Mucopolysaccharidoses (MPS)
- Deficiency of enzymes degrading glycosaminoglycans (heparan sulfate, dermatan sulfate, etc.) → accumulate in lysosomes
- Hurler syndrome (MPS I): α-L-iduronidase deficiency → coarse facial features, corneal clouding, organomegaly, intellectual disability, cardiac disease, short stature
8. GLYCOGEN STORAGE DISEASES (Glycogenoses)
All are autosomal recessive. Result from deficiency of enzymes involved in glycogen synthesis or degradation.
| Type | Disease | Deficient Enzyme | Primary Organ | Key Features |
|---|---|---|---|---|
| Type I (Hepatic) | Von Gierke disease | Glucose-6-phosphatase | Liver + kidney | Hepatomegaly, renomegaly, severe hypoglycemia (fasting), hyperlipidemia, hyperuricemia (gout), bleeding tendency |
| Type II (Generalized) | Pompe disease | Lysosomal acid maltase (acid α-glucosidase) | Heart + muscle | Massive cardiomegaly, hypotonia, cardiorespiratory failure before age 2; adult form = chronic myopathy |
| Type V (Myopathic) | McArdle disease | Muscle phosphorylase | Skeletal muscle | Exercise-induced painful muscle cramps, myoglobinuria; no rise in venous lactate after exercise; adult onset |
9. SINGLE-GENE DISORDERS WITH ATYPICAL (NON-CLASSICAL) PATTERNS OF INHERITANCE
Three groups fall outside standard Mendelian rules:
9A. Triplet Repeat Mutations — Fragile X Syndrome (FXS)
- Gene: FMR1 (Familial Mental Retardation 1) on the X chromosome
- Mutation: Expansion of CGG trinucleotide repeats in the 5' UTR
- Frequency: 1 in 1,550 males; 1 in 8,000 females
- Most common genetic cause of intellectual disability in males; second most common overall cause after Down syndrome
Repeat sizes:
| Category | CGG Repeats | Effect |
|---|---|---|
| Normal | ~5–54 (avg. 29) | Normal FMRP production |
| Premutation | 55–200 | Carrier state; no FXS, but risk of FXTAS/FXPOI |
| Full mutation | 200–4,000 | DNA hypermethylation → gene silencing → no FMRP → FXS |
Pathogenesis: Full mutation → 5' CpG island hypermethylation → silencing of FMR1 → absent FMRP (an RNA-binding protein that normally shuttles mRNAs to axons/dendrites and regulates synaptic translation) → excess mRNA translation at synapses → intellectual disability
Clinical features of FXS (males):
- Severe intellectual disability
- Long face, large mandible, large everted ears, macroorchidism (present in >90% post-pubertal males — most consistent physical finding)
- High arched palate, mitral valve prolapse, hyperextensible joints
- Epilepsy (30%), aggressive behavior (90%), autism spectrum disorder, anxiety/ADHD
Atypical inheritance features:
- Carrier males (premutation): Clinically normal but transmit through phenotypically normal daughters to affected grandchildren ("normal transmitting males")
- Affected females: ~20% of carrier females are affected (much higher than typical X-linked recessive)
- Anticipation: Disease worsens over successive generations as premutation expands to full mutation during oogenesis (NOT spermatogenesis)
- FXTAS (Fragile X-associated Tremor/Ataxia Syndrome): Premutation carrier males/females → late-onset cerebellar ataxia and tremor
- FXPOI (Fragile X-associated Primary Ovarian Insufficiency): Premutation carrier females → premature ovarian failure
9B. Mitochondrial Gene Mutations
- Mitochondria have their own circular DNA (~16,500 bp, 37 genes) encoding 13 proteins of the oxidative phosphorylation chain, 22 tRNAs, 2 rRNAs
- Maternal inheritance: Mitochondria are exclusively maternally inherited (sperm contribute no cytoplasm to the fertilized ovum)
- Heteroplasmy: Cells may have mixtures of mutant and normal mitochondria; disease severity correlates with proportion of mutant mitochondria
- Threshold effect: Minimum number of mutant mitochondria required to impair cell function
- Primarily affect high-energy demanding tissues: CNS, skeletal muscle, cardiac muscle, kidney, liver
Example — MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, Stroke-like episodes): Mutation in mt-tRNA gene (commonly m.3243A>G)
Key features of mitochondrial diseases: Weakness, myopathy, lactic acidosis, encephalopathy, deafness, retinopathy — variable combination depending on mutation and heteroplasmy level
9C. Genomic Imprinting — Prader-Willi and Angelman Syndromes
Imprinting: Certain genes are expressed from only one parental allele (the other is silenced by epigenetic marks — DNA methylation). This is established in the germline and maintained throughout life.
| Feature | Prader-Willi Syndrome (PWS) | Angelman Syndrome (AS) |
|---|---|---|
| Chromosomal region | 15q11–q13 | 15q11–q13 |
| Deleted/silenced allele | Paternal chromosome 15 (paternal genes at 15q11 not expressed) | Maternal chromosome 15 (maternal UBE3A not expressed) |
| Key gene silenced | SNRPN, NDN and others (paternal copies) | UBE3A (maternal copy) |
| Mechanism | Deletion of 15q11 from father (70%) OR maternal uniparental disomy (UPD) 25% | Deletion of 15q11 from mother (70%) OR paternal UPD 5% |
| Clinical features | Hypotonia at birth, hyperphagia, obesity, short stature, hypogonadism, mild intellectual disability | Severe intellectual disability, seizures, ataxic gait, inappropriate laughter ("happy puppet"), absence of speech |
| Mnemonic | Prader-Willi = Weight (obese), Word-eating (hyperphagia) | Angelman = Attempt at gait (ataxia), Abnormal laughter |
10. HIGH-YIELD SUMMARY TABLE
| Disease | Inheritance | Gene/Protein | Key Mechanism | Classic Exam Finding |
|---|---|---|---|---|
| Marfan | AD | FBN1/Fibrillin | ↑TGF-β, defective microfibrils | Aortic dissection, ectopia lentis, arachnodactyly |
| Familial hypercholesterolemia | AD | LDL receptor | ↑LDL, unchecked cholesterol synthesis | Tendon xanthomas, premature MI |
| Cystic fibrosis | AR | CFTR/Cl⁻ channel | Thick mucus → infection → bronchiectasis | Sweat Cl⁻ >60, meconium ileus |
| PKU | AR | PAH | ↑Phenylalanine → brain damage | Musty odor, intellectual disability, fair skin |
| Tay-Sachs | AR | HexA | GM2 ganglioside in neurons | Cherry-red macula, no organomegaly |
| Gaucher Type 1 | AR | Glucocerebrosidase | Glucocerebroside in macrophages | "Wrinkled tissue paper" cells, hepatosplenomegaly |
| Fragile X | X-linked (atypical) | FMR1/FMRP | CGG expansion → methylation → gene silencing | Macroorchidism, anticipation, carrier males |
| Prader-Willi | Imprinting | 15q11 (paternal) | Loss of paternal expression | Obesity, hyperphagia, hypotonia |
| Angelman | Imprinting | UBE3A (maternal) | Loss of maternal UBE3A | Severe ID, seizures, ataxia, laughter |
| Von Gierke | AR | G6Pase | Hypoglycemia, glycogen in liver/kidney | Massive hepatomegaly, hypoglycemia |
| Pompe | AR | Acid maltase | Glycogen in lysosomes of heart/muscle | Cardiomegaly + hypotonia → death <2 yr |
11. EXAM HIGH-YIELD MNEMONICS
- Marfan: MALE = Marfan, Aorta (dissection), Lens (ectopia lentis), Extremities (arachnodactyly)
- CF: "5-7" — CFTR gene on chromosome 7q31, ΔF508 most common mutation
- PKU: Pale, musty smell, Phenylalanine → no tyrosine → no melanin
- Fragile X: FAME = Fragile X, Anticipation, Macroorchidism, Ears (large), premutation in females → expansion in oogenesis
- Gaucher: Gaucher = Glucose-cerebroside in Great big macrophages with "Grinkled tissue paper" look
- Tay-Sachs: Tay = Twice (hexosaminidase A has 2 subunits α and β; Tay-Sachs = α deficient; Sandhoff = both α and β deficient)
- Prader-Willi vs Angelman: "Paternal deletion → Prader-Willi"; "maternal deletion → Angel (from mom)"
— Robbins & Kumar Basic Pathology (Robbins Pathology), 10th ed., Chapter 4; Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 4
This is a shared conversation. Sign in to Orris to start your own chat.