Heme catabolism and fate of bilirubin with diseases biochemistry
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the complete answer.
Heme Catabolism and Fate of Bilirubin - Biochemistry
Sources: Harper's Illustrated Biochemistry (32nd Ed), Lippincott's Biochemistry (8th Ed), Robbins Pathology, Goldman-Cecil Medicine, Sleisenger & Fordtran's GI & Liver Disease
1. Overview
Heme catabolism is the process by which the iron-porphyrin moiety of hemoproteins is dismantled, ultimately producing bilirubin. A normal 70-kg adult destroys ~200 billion erythrocytes per day and turns over ~6 g of hemoglobin daily. This generates 250-350 mg of bilirubin per day (roughly 4 mg/kg/day).
Sources of bilirubin:
- 70-80% from hemoglobin in senescent red blood cells (sequestered by reticuloendothelial cells)
- 20-30% from other heme proteins (myoglobin, cytochromes P450, catalase, peroxidase) mainly in hepatocytes
- Minor fraction from ineffective erythropoiesis (premature destruction of newly formed erythrocytes)
2. Step-by-Step Heme Catabolism
Step 1 - Heme to Biliverdin (in Reticuloendothelial Cells)
The key diagram from Harper's Biochemistry:
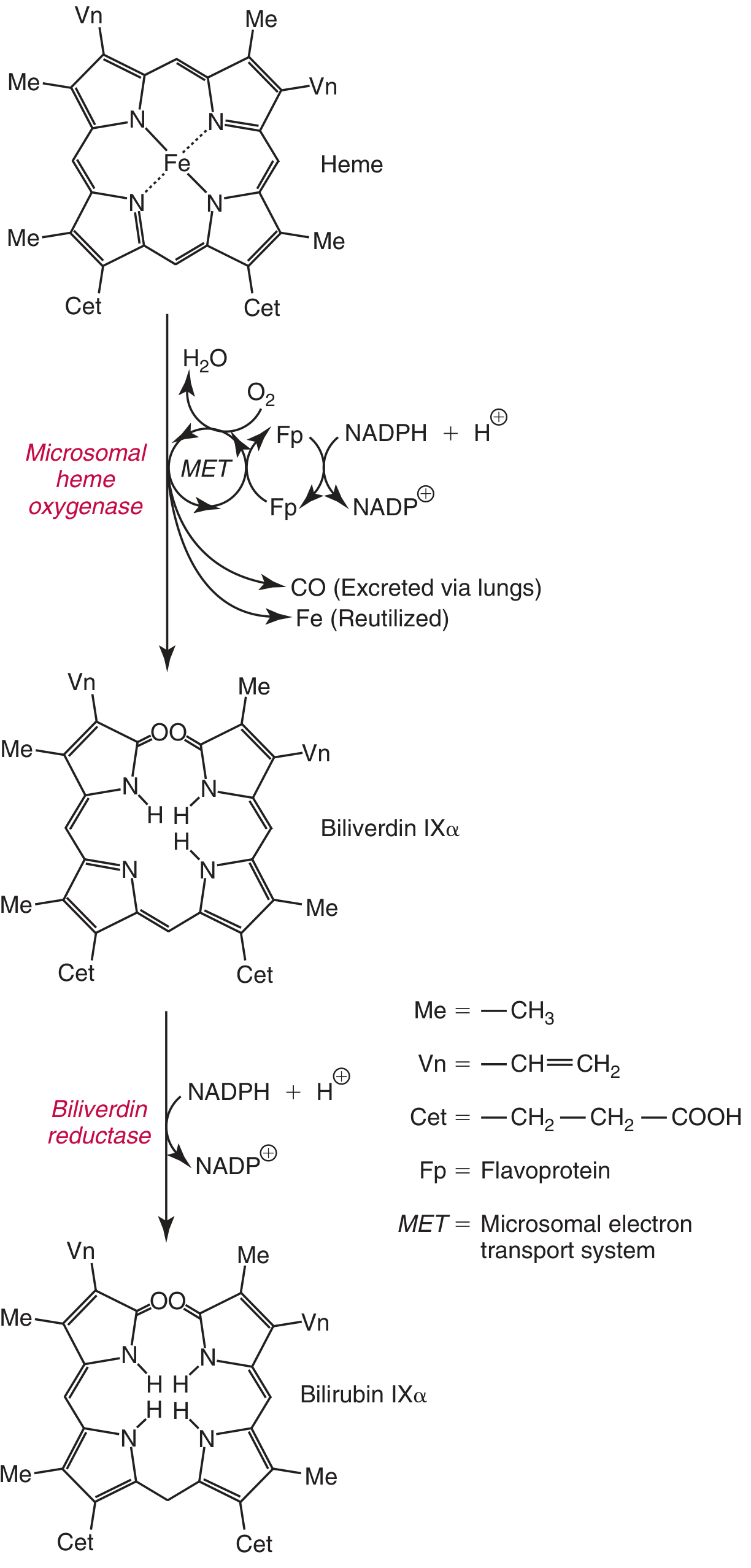
The reaction occurs in the microsomal fraction of reticuloendothelial cells (spleen, bone marrow, liver Kupffer cells) via heme oxygenase (HO), EC 1.14.18.18:
Fe³⁺-Heme + 3 O₂ + 7 e⁻ → Biliverdin + CO + Fe³⁺
Key features:
- Heme oxygenase is substrate-inducible (heme both induces and acts as substrate)
- The iron in heme is typically already oxidized to ferric form (hemin) before reaching HO
- 3 moles of O₂ are consumed; electrons are provided by NADPH via cytochrome P450 reductase
- The heme ring is opened at the α-methene bridge, releasing a green pigment - biliverdin IXα
- CO is released (has signaling and anti-inflammatory roles; excreted via lungs)
- Fe³⁺ is released and reutilized (enters the iron pool)
- Two isoforms: HO-1 (inducible, ER, ubiquitous) and HO-2 (constitutive, mitochondria, in selected tissues including hepatocytes)
Step 2 - Biliverdin to Bilirubin
Biliverdin is then reduced by biliverdin reductase (BVRA), EC 1.3.1.24:
Biliverdin + NADPH + H⁺ → Bilirubin IXα + NADP⁺
- The central methylene bridge of biliverdin is reduced, forming the yellow-orange pigment bilirubin
- This step is unique to mammals (birds and amphibians excrete biliverdin directly)
- Bilirubin also serves as an antioxidant at physiologic concentrations; it cycles bilirubin → biliverdin → bilirubin via BVR, regenerating protective antioxidant capacity
Visual clue: The color changes of a bruise (purple → green → yellow) directly reflect heme → biliverdin → bilirubin conversion in the tissues.
3. Transport to the Liver
Bilirubin at this stage is unconjugated (indirect) bilirubin - nonpolar, sparingly soluble in water, and potentially toxic (can cross the blood-brain barrier).
- Binds noncovalently to serum albumin for transport in the blood
- Albumin has a high-affinity site (~25 mg bilirubin/100 mL plasma) and a low-affinity site
- Certain drugs (salicylates, sulfonamides, rifampin) can displace bilirubin from albumin, increasing free bilirubin and risk of kernicterus in neonates
4. Hepatic Processing of Bilirubin (3 Stages)
Stage 1: Hepatic Uptake
- Bilirubin is removed from albumin at the sinusoidal surface of hepatocytes via a large-capacity, saturable facilitated transport system
- Internalized bilirubin binds to cytosolic ligandin (glutathione S-transferase), which prevents it from re-entering the bloodstream
- This uptake step is not normally rate-limiting
Stage 2: Conjugation with Glucuronic Acid
- Bilirubin is nonpolar and must be made water-soluble for excretion
- Bilirubin UDP-glucuronosyltransferase (UGT1A1) in the ER catalyzes sequential conjugation:
Bilirubin + UDP-glucuronate → Bilirubin monoglucuronide + UDPBilirubin monoglucuronide + UDP-glucuronate → Bilirubin diglucuronide + UDP
- The diglucuronide (conjugated/direct bilirubin) is the predominant form in bile (~80%)
- This transformation makes bilirubin water-soluble, non-toxic, and excretable
Stage 3: Secretion into Bile
- Conjugated bilirubin is actively transported into bile canaliculi by MOAT (Multispecific Organic Anion Transporter), also called MRP2 (Multidrug-Resistant Protein 2) - a member of the ATP-binding cassette (ABC) transporter family
- This is the rate-limiting step for overall hepatic bilirubin metabolism
- Both conjugation and excretion are induced by the same drugs (e.g., phenobarbital)
- Deficiency in MRP2 → Dubin-Johnson syndrome (conjugated hyperbilirubinemia)
5. Fate in the Intestine - The Enterohepatic Circulation
Once conjugated bilirubin reaches the terminal ileum and colon:
- Bacterial deconjugation cleaves glucuronate
- Bacterial reduction produces urobilinogen (colorless)
- Most urobilinogen is further oxidized by bacteria to stercobilin → excreted in feces (gives stool its characteristic brown color)
- A small fraction of urobilinogen is reabsorbed from the gut into portal blood:
- Most is re-extracted by the liver and re-excreted in bile (enterohepatic circulation)
- A small amount escapes into the systemic circulation and is excreted by the kidneys as urobilin (gives urine its yellow color)
6. δ-Bilirubin
When conjugated bilirubin remains elevated for a prolonged period (e.g., chronic biliary obstruction), a fraction covalently binds to albumin - this is called δ-bilirubin (delta-bilirubin). It has a longer half-life (same as albumin, ~21 days) and explains why some patients remain jaundiced even after the obstruction is relieved and standard bilirubin levels normalize.
7. Laboratory Parameters in Jaundice
From Harper's Biochemistry (Table 31-4):
| Condition | Serum Bilirubin | Urine Urobilinogen | Urine Bilirubin | Fecal Urobilinogen |
|---|---|---|---|---|
| Normal | Direct: 0.1-0.4 mg/dL; Indirect: 0.2-0.7 mg/dL | 0-4 mg/24h | Absent | 40-280 mg/24h |
| Hemolytic anemia | ↑ Indirect | Increased | Absent | Increased |
| Hepatitis | ↑ Direct + Indirect | Decreased (if micro-obstruction) | Present (if micro-obstruction) | Decreased |
| Obstructive jaundice | ↑ Direct | Absent | Present | Trace to absent |
Key diagnostic rule: Bilirubin appears in urine only when it is in the conjugated (direct) form, since unconjugated bilirubin is bound to albumin and cannot be filtered by the glomerulus.
8. Diseases of Bilirubin Metabolism
Classification by Site
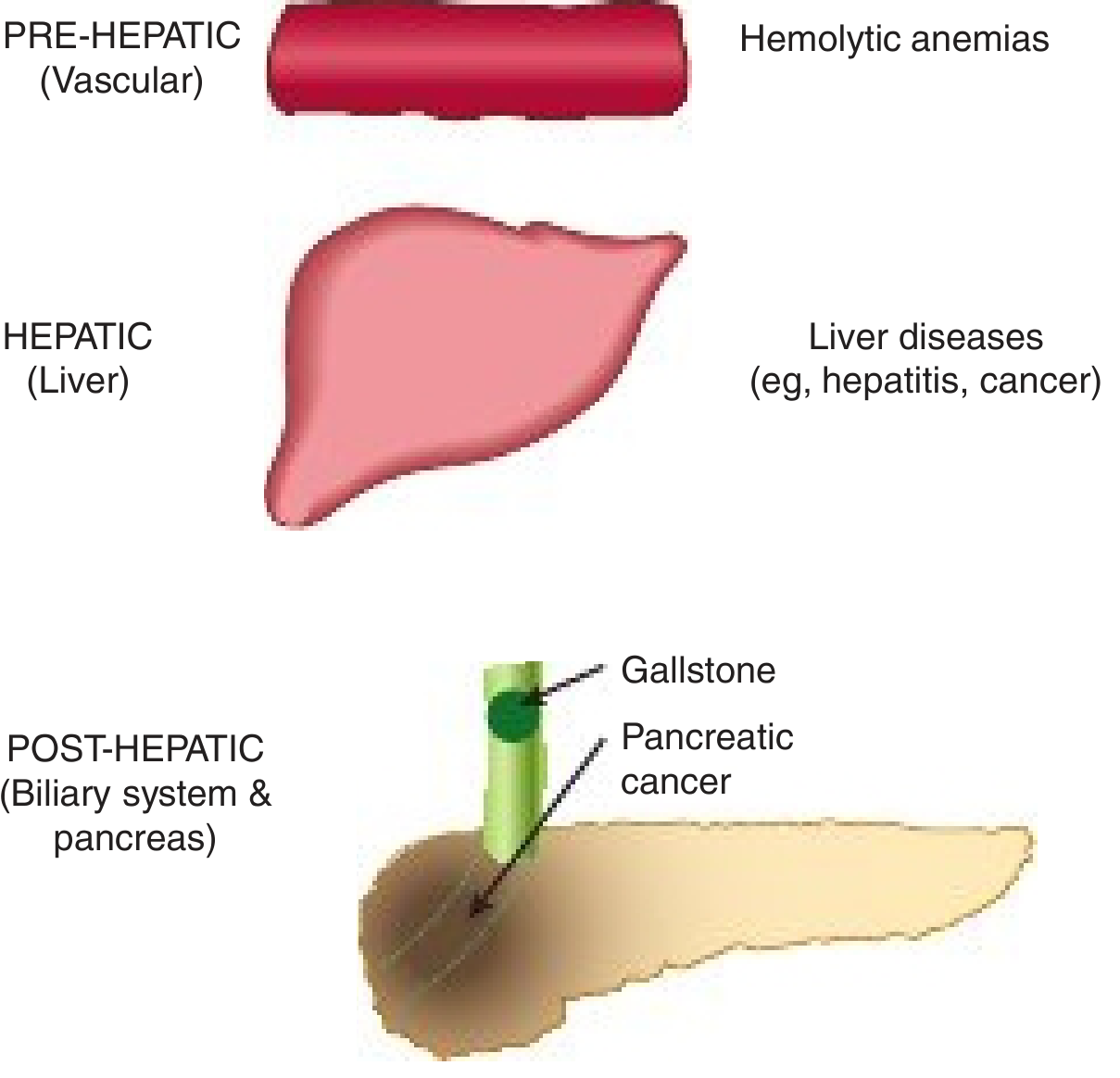
A. UNCONJUGATED HYPERBILIRUBINEMIAS
1. Hemolytic (Pre-hepatic) Jaundice
- Excessive RBC destruction overwhelms the liver's conjugation capacity
- ↑ Indirect bilirubin; bilirubin usually modest (<4 mg/dL) because the healthy liver has large reserve capacity
- No bilirubinuria (unconjugated bilirubin cannot pass glomerulus)
- ↑ Urobilinogen in urine and stool
- Examples: sickle cell anemia, thalassemia, G6PD deficiency, autoimmune hemolysis
2. Neonatal "Physiologic" Jaundice
- Results from accelerated hemolysis + immature UGT1A1 activity (low enzyme + low UDP-glucuronate synthesis)
- UGT1A1 levels are low at birth; do not reach adult levels until 3-4 months
- Breast milk jaundice: bilirubin-deconjugating enzymes in breast milk may worsen hyperbilirubinemia
- Risk: unconjugated bilirubin >20-25 mg/dL can cross the blood-brain barrier → kernicterus (toxic encephalopathy causing mental retardation)
- Treatment: phototherapy with blue light (converts bilirubin to water-soluble photoisomers excreted in urine/bile without needing conjugation); phenobarbital (induces UGT1A1)
3. Gilbert Syndrome
- Most common hereditary hyperbilirubinemia
- Autosomal recessive - homozygous polymorphism in the UGT1A1 promoter (UGT1A1*28: extra TA repeat in the TATA box → ↓ UGT1A1 transcription → ~30% residual UGT1A1 activity)
- Allele frequencies: 42% Black, 30% White, 10% Asian populations
- Mild unconjugated hyperbilirubinemia (<3 mg/dL); increases 2-3 fold with fasting, stress, illness
- Clinically benign - no liver damage
- Clinical relevance: Irinotecan (CPT-11) toxicity - active metabolite SN-38 is glucuronidated by UGT1A1; patients with Gilbert's get severe diarrhea and myelosuppression. Also affects raloxifene metabolism.
- Phenobarbital normalizes bilirubin
4. Crigler-Najjar Syndrome Type I
- Complete absence of UGT1A1 activity (autosomal recessive; nonsense mutations in UGT1A1 gene)
- Severe congenital jaundice: bilirubin >20 mg/dL
- Fatal within the first 15 months if untreated (kernicterus)
- Phenobarbital has no effect (no enzyme to induce)
- Treatment: prolonged phototherapy (12-16 hrs/day) + ultimately liver transplantation
5. Crigler-Najjar Syndrome Type II (Arias Syndrome)
- Reduced (but not absent) UGT1A1 activity
- Serum bilirubin <20 mg/dL
- More benign; responds to phenobarbital (which induces residual enzyme)
6. Toxic Hyperbilirubinemia
- Hepatocyte damage → impaired conjugation
- Causes: chloroform, CCl₄, acetaminophen overdose, hepatitis viruses, cirrhosis, Amanita mushroom poisoning, certain drugs (isoniazid, diclofenac)
- Mostly unconjugated initially; mixed (conjugated + unconjugated) with cell necrosis
B. CONJUGATED (DIRECT) HYPERBILIRUBINEMIAS
7. Dubin-Johnson Syndrome
- Autosomal recessive - mutation in MRP2 gene (canalicular transporter for organic anions including conjugated bilirubin)
- Impaired secretion of conjugated bilirubin into bile canaliculi
- Conjugated hyperbilirubinemia (mild, <7 mg/dL); usually presents in childhood/adulthood
- Striking feature: deposition of brown-black melanin-like pigment in hepatocytes → can blacken the liver grossly
- Clinically benign; normal life expectancy
- Total urinary coproporphyrin normal but ratio of isomers I/III is reversed (>80% isomer I vs normally <25%)
8. Rotor Syndrome
- Autosomal recessive - mutation in SLCO1B1/SLCO1B3 genes (organic anion transporting polypeptides OATP1B1/1B3) - defective reuptake of bilirubin conjugates secreted into the space of Disse back into hepatocytes
- Similar to Dubin-Johnson but no liver pigment deposition
- Total urinary coproporphyrin elevated (isomers I + III both high)
- Clinically benign
9. Obstructive (Post-hepatic/Cholestatic) Jaundice
- Blockage of hepatic or common bile duct prevents excretion of conjugated bilirubin
- Most common causes: gallstone in CBD or carcinoma of the head of the pancreas
- Conjugated bilirubin regurgitates into hepatic veins and lymphatics → enters blood and urine (choluria/bilirubinuria)
- Pale/clay-colored stools (acholic - no stercobilin reaches gut)
- No urobilinogen in urine (no bilirubin reaches the intestine)
- ↑ Alkaline phosphatase, ↑ GGT (cholestatic markers)
- Persistent obstruction → biliary cirrhosis, ascending cholangitis
10. Hepatocellular Disease (Hepatitis/Cirrhosis)
- Mixed conjugated + unconjugated hyperbilirubinemia
- Impaired uptake, conjugation, AND secretion
- Both types of bilirubin elevated; ↑ ALT, ↑ AST (hepatocellular injury markers)
- Urobilinogen may be decreased (if micro-obstruction of bile ductules occurs)
9. Summary Table: Hereditary Hyperbilirubinemias
| Syndrome | Defect | Type | Bilirubin | Severity | Treatment |
|---|---|---|---|---|---|
| Gilbert | ↓ UGT1A1 expression (UGT1A1*28) | Unconjugated | <3 mg/dL | Benign | None needed |
| Crigler-Najjar I | Absent UGT1A1 | Unconjugated | >20 mg/dL | Fatal | Phototherapy + liver Tx |
| Crigler-Najjar II | Reduced UGT1A1 | Unconjugated | <20 mg/dL | Moderate | Phenobarbital |
| Dubin-Johnson | Absent MRP2 (canalicular) | Conjugated | Mild | Benign | None |
| Rotor | Absent OATP1B1/1B3 | Conjugated | Mild | Benign | None |
10. Key Biochemical Points for Exams
- Heme oxygenase is microsomal, substrate-inducible; uses 3 O₂ + 7 electrons (NADPH)
- CO is a byproduct (excreted via lungs) - also a signaling molecule
- Biliverdin reductase uses NADPH; mammals unique in this reduction step
- 1 g hemoglobin → ~35 mg bilirubin
- Ligandin (glutathione S-transferase) - intrahepatic bilirubin binding protein
- UGT1A1 - key conjugating enzyme; inducible by phenobarbital
- MRP2 - canalicular ABC transporter; rate-limiting step for bilirubin excretion
- Conjugated bilirubin is water-soluble, appears in urine; unconjugated does not (albumin-bound)
- Jaundice clinically detectable when bilirubin >3-4 mg/dL
- Kernicterus risk when unconjugated bilirubin >20-25 mg/dL (crosses BBB)
- Harper's Illustrated Biochemistry, 32nd Ed, pp. 333-336
- Lippincott's Illustrated Reviews: Biochemistry, 8th Ed, pp. 796-798
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 976-982
- Goldman-Cecil Medicine, 2-Volume Set, pp. 1551-1555
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease, pp. 2684-2692
This is a shared conversation. Sign in to Orris to start your own chat.