Sjogrens syndrome ILD full details for md exams 15marks answer from fishman and murray and ats and ers guidelines just from these only
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed. Let me compose the comprehensive 15-mark MD exam answer.
Sjögren Syndrome - Interstitial Lung Disease (ILD)
A Comprehensive MD Examination Answer (15 Marks)
Sources: Fishman's Pulmonary Diseases and Disorders (2-Vol Set), Murray & Nadel's Textbook of Respiratory Medicine (2-Vol Set)
I. INTRODUCTION AND OVERVIEW
Sjögren syndrome (SS) is an autoimmune exocrinopathy characterized by lymphocytic infiltration of the lacrimal and salivary glands, producing the clinical triad of xerophthalmia, xerostomia, and polyarthritis. Two forms exist:
- Primary Sjögren syndrome (pSS): occurs in isolation, without another connective tissue disease (CTD)
- Secondary Sjögren syndrome (sSS): associated with another CTD, most frequently rheumatoid arthritis (RA), but also systemic sclerosis (SSc) and SLE
A strong female predominance (90%) exists. Serologically, rheumatoid factor (RF) is positive in ~95%, antinuclear antibodies show a speckled pattern in ~80%, and anti-SSA (Ro) and anti-SSB (La) antibodies are specific for the primary form.
- Fishman's Pulmonary Diseases and Disorders, Ch. 58
II. EPIDEMIOLOGY AND RISK FACTORS FOR PULMONARY INVOLVEMENT
-
Annual incidence of primary SS: 6.9 per 100,000; prevalence 60.8 per 100,000 (Murray & Nadel, Ch. 92)
-
Presents at two age peaks: 20-30 years and >50 years; predominantly in women
-
Genetic predisposition includes familial clustering and association with HLA-Dw2 and HLA-Dw3
-
Clinically significant ILD develops in 8-27% of patients with SS
-
ILD is more common in older, male patients with a history of smoking and positive ANA testing
-
ILD tends to develop early in the disease course, with a cumulative incidence of 10% at 1 year after diagnosis of systemic disease
-
Objective evidence of pulmonary abnormalities is present in approximately one-fourth of cases with primary SS
-
Murray & Nadel's Textbook of Respiratory Medicine, Ch. 92
III. PATHOGENESIS AND HISTOPATHOLOGY
The unifying mechanism in pSS is lymphocytic infiltration - the same process that destroys lacrimal and salivary glands is replicated in the lung parenchyma and airways.
Key Histologic Patterns in Sjögren Syndrome-ILD:
| Pattern | Description | Frequency in SS |
|---|---|---|
| LIP (Lymphocytic Interstitial Pneumonia) | Monotonous infiltration of interstitium by mature lymphocytes forming germinal centers; angiogenic distribution; macrophagic giant cells, granuloma formation, and amyloid deposition | Most characteristic of primary SS |
| NSIP (Non-specific Interstitial Pneumonitis) | Lymphoplasmacytic infiltration with varying collagen deposition; temporally/spatially homogeneous | Most common pattern overall - 45.5% of biopsies |
| UIP (Usual Interstitial Pneumonitis) | Varying mononuclear infiltration and fibroblastic proliferation leading to collagen deposition; honeycomb lung in advanced cases | Uncommon in primary SS; more frequent in secondary SS |
| OP (Organizing Pneumonia) | Intra-alveolar fibroblastic proliferation (Masson bodies); inflammatory polyps in bronchioles | Less common than in RA or PM |
| Pseudolymphoma | Tumor-like mass; considered a localized form of LIP | Premalignant lesion |
| Pulmonary Lymphoma | MALToma most common | Prevalence increased 40-50 fold |
Important distinction (Fishman, Ch. 58): ILD occurs more commonly in secondary SS and most likely represents a complication of the associated CTD. The histologic pattern in secondary SS mimics that seen in RA, with NSIP, UIP, and organizing pneumonia reported. UIP is uncommon in primary SS.
Histology of LIP in detail (Fishman, Fig. 58-2): Dense lymphocytic infiltrate broadening the interstitium with lymphoid follicle formation. Features include:
- Monotonous mature lymphocyte infiltration
- Germinal center formation within the interstitium
- Angiogenic distribution
- Macrophagic giant cells and granuloma formation
- Amyloid deposition (in some cases)
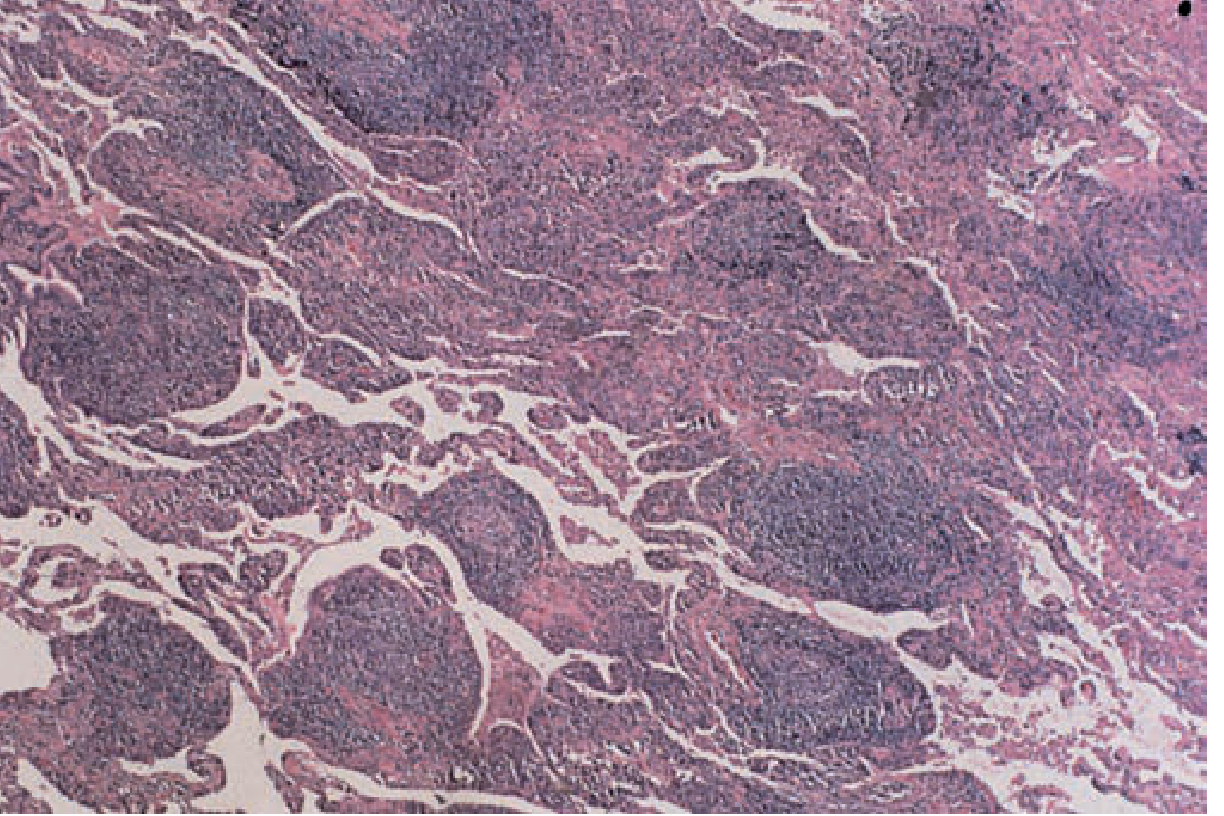
IV. PULMONARY MANIFESTATIONS - SPECTRUM OF DISEASE
Lung involvement in SS falls into two major categories: (1) ILD/diffuse lung disease and (2) tracheobronchial disease. Both result from the same underlying lymphocytic infiltration targeting different compartments.
A. INTERSTITIAL LUNG DISEASE (Primary Form of SS)
Clinical Presentation
- Nonproductive cough, dyspnea on exertion, or asymptomatic radiographic abnormalities
- Crackles on auscultation
- Reticular or nodular opacities on chest radiography
- Restrictive pattern of functional impairment on PFTs with reduced DLCO
- Sicca symptoms (dry eyes, dry mouth) - a key diagnostic clue in any patient presenting with ILD
Lymphocytic Interstitial Pneumonia (LIP) - The Hallmark of Primary SS-ILD
LIP is characterized by:
- Diffuse lymphocytic infiltrate, with or without histiocytes and multinucleated giant cells
- Most prominent around bronchioles
- BAL lymphocytosis is characteristic
- Mixed alveolar AND interstitial infiltrates on radiology (because lymphocytes infiltrate alveolar spaces as well as interstitium)
- In a subset of patients, variably sized cystic lesions with associated ground-glass opacification may be the only radiographic abnormality
Radiologic hallmark of LIP in SS: Multiple cysts of varying sizes scattered throughout both lungs (see image below)
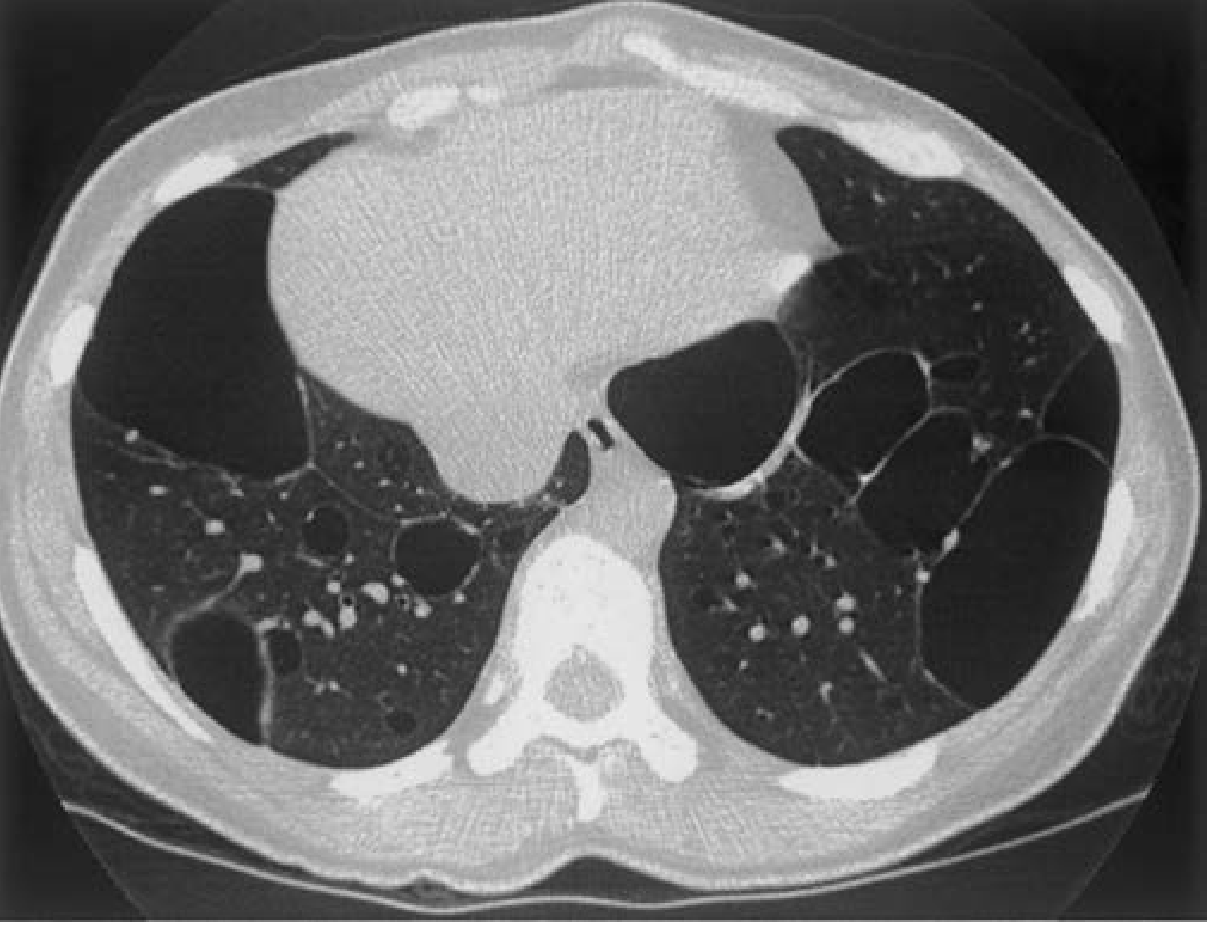
Pseudolymphoma
- A tumor-like proliferation appearing as single or multiple masses on chest radiograph
- Difficult to distinguish from malignant lymphoma
- Considered a localized form of LIP; regarded as a premalignant lesion
- When associated with a monoclonal gammopathy, malignant transformation to lymphoma is suggested
Risk of Lymphomatous Transformation
- Both LIP and pseudolymphoma have potential for lymphomatous conversion - this is clinically critical
- The development of pleural effusions or hilar/mediastinal adenopathy should trigger urgent investigation for malignant transformation to lymphoma
- The prevalence of lymphoma is increased 40- to 50-fold in Sjögren syndrome (Murray & Nadel, Ch. 92)
- Pulmonary lymphoma has a highly variable clinical and radiographic presentation: diffuse interstitial involvement to discrete (often perihilar) masses
B. TRACHEOBRONCHIAL DISEASE
Mechanism
Lymphocytic infiltration and destruction of airway mucus glands result in desiccation of the tracheobronchial tree (xerotrachea).
Clinical Features
- Xerotrachea (atrophy of tracheobronchial mucous glands with lymphoplasmacytic infiltrate): develops in up to 25% of patients with pSS; presents as a relentless dry cough with endobronchial inflammation at bronchoscopy
- Hoarseness, cough, inspissation of secretions leading to luminal obstruction, atelectasis, recurrent pneumonias
- Bronchiectasis
- Follicular bronchiolitis (high incidence) causing obstructive ventilatory dysfunction
- Obliterative/constrictive bronchiolitis and bronchiolectasis
- Small airway disease: Aerosol penetration from central airways to the periphery is reduced, indicating small airway obstruction; abnormalities demonstrated at low lung volumes
- Bronchial hyperresponsiveness in 40-60% of patients with primary and secondary SS
- Subclinical bronchiolitis is common; lymphocytic infiltration more prominent around small bronchioles
Asthma and COPD Association (Murray & Nadel)
- The incidence of asthma was 1.6-fold higher and COPD 1.4-fold higher in SS patients compared to matched controls
- Notably, inhaled anti-inflammatories including inhaled budesonide and inhaled cromoglycate appear to have no effect on bronchial hyperreactivity in SS
V. INVESTIGATIONS
A. Laboratory
- Anti-SSA (Ro) and anti-SSB (La) antibodies - most specific for primary SS-ILD
- RF positive ~95%; ANA positive ~80% (speckled pattern)
- Serum protein electrophoresis - to detect monoclonal gammopathy (risk of lymphoma)
B. Pulmonary Function Tests
- Restrictive pattern: reduced TLC, reduced FVC
- Reduced DLCO (reflects parenchymal involvement)
- Obstructive pattern or mixed when airways are predominantly involved
- Small airway dysfunction detectable in unselected patients by evaluation of airflow at low lung volumes
C. HRCT Chest - Key Patterns
| SS-ILD Subtype | HRCT Findings |
|---|---|
| LIP | Bilateral ground-glass opacification + multiple thin-walled cysts of varying sizes scattered throughout; cysts are the pathognomonic finding |
| NSIP | Bilateral, symmetric, subpleural ground-glass opacification; traction bronchiectasis; reticulation; lower lobe predominance; peribronchovascular distribution |
| UIP | Bilateral basal, subpleural reticulation; honeycombing; traction bronchiectasis; less GGO |
| Airway disease | Bronchial wall thickening, bronchiectasis, tree-in-bud pattern |
| OP | Consolidation (often migratory), peribronchovascular distribution |
In a CT study of 35 patients with SS, the most prevalent findings were large and/or small airway disease (n=19) and diffuse lung disease (n=12, including 7 with features suggestive of LIP). (Murray & Nadel)
D. BAL
- BAL lymphocytosis is a characteristic finding in SS-ILD, reflecting lymphocytic infiltration
- CD4+ T lymphocyte predominance
E. Tissue Diagnosis
- Transbronchial biopsy can reveal fibrosis or lymphocytic infiltration
- Surgical lung biopsy may be needed to confirm NSIP vs. UIP vs. LIP when clinical and HRCT features are not sufficient
- Salivary gland biopsy (minor lip biopsy) can confirm SS
VI. DIAGNOSIS
The diagnosis of SS-ILD rests on:
- Establishing Sjögren syndrome (clinical features + serology + biopsy + Schirmer test + rose bengal staining)
- Characterizing the lung involvement (HRCT pattern + PFTs + BAL)
- Distinguishing primary from secondary SS - critical for management (secondary SS-ILD reflects the underlying CTD's pattern)
- Ruling out drug-induced lung disease and opportunistic infections (important in the differential, especially under immunosuppression) (Murray & Nadel, Ch. 92)
Sicca symptoms in any ILD patient should prompt investigation for subclinical SS - patients with confirmed NSIP pattern on HRCT or biopsy should always be evaluated for an underlying CTD.
VII. PROGNOSIS
- The 5-year survival rate of patients with ILD associated with Sjögren syndrome was 84% (Murray & Nadel, Ch. 92)
- When ILD is present, it is associated with a poorer quality of life and increased mortality
- LIP generally has a better prognosis than UIP
- LIP is responsive to anti-inflammatory therapy in the majority of patients, though a subset progresses to fibrotic lung disease with honeycomb change
- NSIP pattern generally has better prognosis than UIP pattern
- Progression to lymphoma confers a significantly worsened prognosis
VIII. TREATMENT
Treatment is guided by the predominant histologic pattern and severity:
A. Corticosteroids
- First-line treatment for LIP, OP, and NSIP in SS
- LIP is responsive to anti-inflammatory agents such as corticosteroids (Fishman, Ch. 58)
- Pseudolymphoma often responds well to corticosteroid therapy; however, some patients progress to pulmonary lymphoma
- OP: response to corticosteroid therapy is usual but not invariable
B. Cytotoxic/Immunosuppressive Agents
- When corticosteroids alone are insufficient, azathioprine or cyclophosphamide may be required (though remains of unproven benefit for LIP specifically)
- Cyclosporine has been recommended as an additional agent in corticosteroid-resistant cases
C. Biologic Therapy
- Rituximab (anti-CD20 monoclonal antibody):
- May be beneficial in selected patients with primary SS-ILD
- Further controlled studies are required to better assess efficacy
- Anecdotal reports of response in LIP
- A retrospective cohort study showed benefit in primary SS with ILD (Fishman, Ch. 58 references)
D. Monitoring
- Regular HRCT monitoring for disease progression or transformation
- Serial PFTs (FVC, DLCO) to track decline
- Vigilance for signs of lymphomatous transformation: new pleural effusion, hilar/mediastinal adenopathy, monoclonal gammopathy
IX. SUMMARY TABLE - SS-ILD AT A GLANCE
| Feature | Primary SS-ILD | Secondary SS-ILD |
|---|---|---|
| Typical ILD pattern | LIP (most characteristic) | NSIP, UIP, OP (mimics RA) |
| UIP frequency | Uncommon | More common |
| Mechanism | Direct lymphocytic infiltration | Complication of associated CTD |
| HRCT hallmark | Bilateral cysts + GGO (LIP) | Ground-glass, reticulation (NSIP) |
| Lymphoma risk | Increased 40-50 fold | Lower |
| Treatment | Corticosteroids, ± azathioprine, rituximab | Treat underlying CTD + corticosteroids |
| Prognosis | 5-year survival ~84% | Depends on underlying CTD |
X. KEY EXAMINATION POINTS TO REMEMBER
- LIP is the most characteristic ILD pattern of primary SS - bilateral cysts on HRCT are pathognomonic
- NSIP is the most common overall ILD pattern in SS (45.5% of biopsies)
- UIP is uncommon in primary SS but occurs in secondary SS mimicking RA pattern
- Both LIP and pseudolymphoma have potential for lymphomatous conversion - 40-50x increased lymphoma risk
- BAL lymphocytosis is the characteristic BAL finding
- Bronchial hyperresponsiveness in 40-60% of SS patients - but inhaled budesonide/cromoglycate are ineffective
- Xerotrachea (up to 25% of pSS) - relentless dry cough with endobronchial inflammation
- Follicular bronchiolitis is the cause of obstructive physiology in SS
- Sicca symptoms in any ILD patient should trigger SS workup
- Rituximab is the emerging biologic option for refractory SS-ILD
- New-onset pleural effusion or mediastinal adenopathy in SS = suspect lymphoma until proven otherwise
- 5-year survival ~84% for SS-ILD overall
Sources: Fishman's Pulmonary Diseases and Disorders, 2-Volume Set (Ch. 58: Pulmonary Parenchymal and Vascular Pathology in the Collagen Vascular Diseases); Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set (Ch. 92: Connective Tissue Diseases)
This is a shared conversation. Sign in to Orris to start your own chat.