FSHD & LGMD
facioscapulohumeral muscular dystrophy AND limb girdle muscular dystrophy
Facioscapulohumeral Muscular Dystrophy (FSHD) & Limb-Girdle Muscular Dystrophy (LGMD)
FSHD
Overview & Epidemiology
- FSHD1 (~95%): deletions of tandem 3.3-kb repeat units at 4q35, reducing the D4Z4 repeat array to <35 kb
- FSHD2 (~5%): no deletion; instead, mutations in SMCHD1 (most common), DNMT3B, or LRIF1 genes
Clinical Features
| Region | Specific Findings |
|---|---|
| Face | Inability to smile, whistle, or firmly close eyes; orbicularis oculi + oris involved; masseters/extraocular muscles spared |
| Shoulder | Scapular winging ("angel-wing" appearance), loss of scapular stabilizers, difficulty raising arms; biceps/triceps affected but deltoid relatively spared |
| Arms | Wrist extension > wrist flexion weakness |
| Legs | Anterior compartment weakness → foot drop |
| Pelvis | 20% of patients eventually develop pelvic girdle weakness → wheelchair dependency |
- Ventilatory muscle weakness: ~5%
- Sensorineural hearing loss (increased incidence)
- Coats' disease: retinal telangiectasias, exudation, and retinal detachment
- Heart is generally not involved (distinguishes FSHD from many other dystrophies)
Labs & Investigations
- Serum CK: normal or mildly elevated
- EMG: nonspecific myopathic pattern
- Muscle biopsy: nonspecific dystrophic changes; can show prominent inflammatory infiltrate - may be misdiagnosed as myositis
- Genetic testing: diagnosis confirmed by D4Z4 repeat size analysis (FSHD1) or SMCHD1/DNMT3B/LRIF1 sequencing (FSHD2)
Treatment
- Physical and occupational therapy
- Ankle-foot orthoses (AFO) for foot drop
- Scapular fixation surgery: improves winging and function
- Respiratory monitoring (spirometry) for the 5% with ventilatory involvement
Limb-Girdle Muscular Dystrophy (LGMD)
Overview & Epidemiology
- Progressive proximal (pelvic and shoulder girdle) weakness
- Independent ambulation achieved at some point
- Elevated CK
- Dystrophic features on biopsy or imaging
- At least two unrelated families reported with the same mutation
Classification - ENMC Nomenclature (2018)
- LGMDD (Dominant) + number
- LGMDR (Recessive) + number
| Old Name | New Name | Gene/Protein | Key Feature |
|---|---|---|---|
| LGMD2A | LGMDR1 | CAPN3 / Calpain-3 | Most common; scapular winging; no cardiac/respiratory involvement; common in Southern Europe |
| LGMD2B | LGMDR2 | DYSF / Dysferlin | Calf-predominant initially (Miyoshi myopathy overlap) |
| LGMD2C-F | LGMDR3-6 | Sarcoglycans (γ, α, β, δ) | Sarcoglycanopathies; can resemble DMD |
| LGMD2I | LGMDR9 | FKRP / Fukutin-related protein | Common in northern Europeans; calf hypertrophy; cardiac + respiratory involvement |
| LGMD2L | LGMDR12 | ANO5 / Anoctamin-5 | ~7% of LGMD in the US; medial calf atrophy; overlaps with dysferlinopathy pattern |
| LGMD1B | LGMDD1 | LMNA / Lamin A/C | Now reclassified under EDMD; cardiac conduction defects prominent |
Note: Laminopathies (old LGMD1B) and myofibrillar myopathies (old LGMD1A/myotilin) are now reclassified out of LGMD into EDMD and MFM groups respectively under the ENMC system.
Clinical Features
- Progressive proximal weakness of pelvic + shoulder girdle muscles
- Often clinically indistinguishable from Duchenne/Becker muscular dystrophy
- Respiratory insufficiency from diaphragm weakness - variable by subtype
- Cardiomyopathy - variable by subtype (prominent in LGMDR9/FKRP, laminopathies)
- Serum CK elevated (often markedly)
- EMG: myopathic
- Muscle biopsy: dystrophic features + immunohistochemistry (sarcoglycans, dysferlin, α-dystroglycan) helps narrow subtype
Diagnosis
Treatment
- No curative therapy for most subtypes
- Pompe disease (acid maltase deficiency, formerly LGMD2V/LGMDR22): enzyme replacement therapy (alglucosidase alfa) is available and effective - the only LGMD subtype with approved disease-modifying therapy
- Physical therapy, respiratory support (NIV when needed), cardiac surveillance and management (ACE inhibitors/beta-blockers for cardiomyopathy)
- Regular cardiac monitoring especially for LGMDR9 (FKRP), laminopathies, and sarcoglycanopathies
FSHD vs LGMD - Key Comparisons
| Feature | FSHD | LGMD |
|---|---|---|
| Pattern | Facial + scapulohumeral + descending | Proximal pelvic + shoulder girdle |
| Inheritance | Autosomal dominant (AD) | AD (LGMDD) or AR (LGMDR); AR more common |
| Genetics | D4Z4 deletion at 4q35 (FSHD1); SMCHD1/DNMT3B/LRIF1 (FSHD2) | Heterogeneous: calpain-3, dysferlin, sarcoglycans, FKRP, anoctamin-5, etc. |
| Facial weakness | Yes - hallmark | No |
| Cardiac | Typically spared | Yes, in several subtypes (FKRP, laminopathies) |
| Respiratory | ~5% | Variable; prominent in some subtypes |
| CK | Normal to mildly elevated | Markedly elevated |
| Hearing loss | Yes (increased incidence) | No |
| Coats' disease | Yes | No |
| Disease-modifying Rx | None (DUX4 trials ongoing) | ERT for Pompe; none for others |
| Scapular winging | Prominent, early | Can occur in calpainopathy; less prominent |
Can u generate a ppt for me on these 2 topics
mkdir -p /home/daytona/workspace/fshd-lgmd-ppt && ls /home/daytona/workspace/fshd-lgmd-ppt/
~/fshd-lgmd-ppt/generate.js
const pptxgen = require("pptxgenjs");
const pres = new pptxgen();
pres.layout = "LAYOUT_16x9";
pres.title = "FSHD & LGMD - Muscular Dystrophies";
pres.author = "Medical Education";
// ─── COLOR PALETTE ───────────────────────────────────────────────
// Deep navy dominant, teal accent, warm coral highlight
const C = {
navyDark: "0D1B2A", // dark bg
navy: "1B2E45", // mid bg
navyMid: "1E3A5F", // card bg
teal: "00BFA6", // accent 1
tealDark: "007D6B", // accent 1 dark
coral: "FF6B6B", // accent 2 / LGMD
sky: "4FC3F7", // light accent
white: "FFFFFF",
offWhite: "E8F0FE",
gray: "B0BEC5",
lightGray: "CFD8DC",
cardBg: "132438",
fshd: "00BFA6", // FSHD = teal
lgmd: "FF6B6B", // LGMD = coral
yellow: "FFD600",
};
// ─── HELPERS ─────────────────────────────────────────────────────
function darkSlide(slide) {
slide.background = { color: C.navyDark };
}
function midSlide(slide) {
slide.background = { color: C.navy };
}
function sectionDivider(slide, label, color, subtitle) {
slide.background = { color: C.navyDark };
// accent bar left
slide.addShape(pres.shapes.RECTANGLE, { x: 0, y: 0, w: 0.18, h: 5.625, fill: { color: color }, line: { type: "none" } });
// big label
slide.addText(label, {
x: 0.4, y: 1.5, w: 9.3, h: 1.6,
fontSize: 52, bold: true, color: C.white, fontFace: "Calibri",
align: "left", valign: "middle"
});
if (subtitle) {
slide.addText(subtitle, {
x: 0.4, y: 3.2, w: 9.3, h: 0.7,
fontSize: 20, color: color, fontFace: "Calibri",
align: "left", bold: false
});
}
// decorative circle
slide.addShape(pres.shapes.OVAL, { x: 8.2, y: 0.4, w: 1.8, h: 1.8, fill: { color: color, transparency: 80 }, line: { type: "none" } });
slide.addShape(pres.shapes.OVAL, { x: 8.5, y: 0.8, w: 1.2, h: 1.2, fill: { color: color, transparency: 50 }, line: { type: "none" } });
}
function slideTitle(slide, title, accent) {
// top bar
slide.addShape(pres.shapes.RECTANGLE, { x: 0, y: 0, w: 10, h: 0.72, fill: { color: C.navyDark }, line: { type: "none" } });
slide.addShape(pres.shapes.RECTANGLE, { x: 0, y: 0.72, w: 10, h: 0.06, fill: { color: accent || C.teal }, line: { type: "none" } });
slide.addText(title, {
x: 0.3, y: 0.05, w: 9.4, h: 0.62,
fontSize: 22, bold: true, color: C.white, fontFace: "Calibri",
align: "left", valign: "middle", margin: 0
});
}
function infoCard(slide, x, y, w, h, title, titleColor, lines) {
slide.addShape(pres.shapes.RECTANGLE, { x, y, w, h, fill: { color: C.cardBg }, line: { color: titleColor, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 8, offset: 3, angle: 135, opacity: 0.25 } });
// card title bar
slide.addShape(pres.shapes.RECTANGLE, { x, y, w, h: 0.38, fill: { color: titleColor, transparency: 15 }, line: { type: "none" } });
slide.addText(title, { x: x + 0.12, y: y + 0.03, w: w - 0.2, h: 0.33, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
// body
const items = lines.map((l, i) => ({
text: l,
options: { bullet: { type: "bullet" }, breakLine: i < lines.length - 1, fontSize: 11.5, color: C.offWhite, fontFace: "Calibri" }
}));
slide.addText(items, { x: x + 0.12, y: y + 0.43, w: w - 0.2, h: h - 0.53, valign: "top", margin: 0 });
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 1 — TITLE SLIDE
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.navyDark };
// gradient-like side accent
s.addShape(pres.shapes.RECTANGLE, { x: 0, y: 0, w: 0.35, h: 5.625, fill: { color: C.teal }, line: { type: "none" } });
s.addShape(pres.shapes.RECTANGLE, { x: 0.35, y: 0, w: 0.12, h: 5.625, fill: { color: C.teal, transparency: 60 }, line: { type: "none" } });
// FSHD pill
s.addShape(pres.shapes.ROUNDED_RECTANGLE, { x: 0.7, y: 0.9, w: 2.2, h: 0.5, fill: { color: C.teal, transparency: 20 }, line: { type: "none" }, rectRadius: 0.12 });
s.addText("FSHD", { x: 0.7, y: 0.9, w: 2.2, h: 0.5, fontSize: 16, bold: true, color: C.white, align: "center", valign: "middle", fontFace: "Calibri", margin: 0 });
// LGMD pill
s.addShape(pres.shapes.ROUNDED_RECTANGLE, { x: 3.1, y: 0.9, w: 2.2, h: 0.5, fill: { color: C.coral, transparency: 20 }, line: { type: "none" }, rectRadius: 0.12 });
s.addText("LGMD", { x: 3.1, y: 0.9, w: 2.2, h: 0.5, fontSize: 16, bold: true, color: C.white, align: "center", valign: "middle", fontFace: "Calibri", margin: 0 });
// main title
s.addText("Muscular Dystrophies", {
x: 0.6, y: 1.55, w: 8.8, h: 1.1,
fontSize: 46, bold: true, color: C.white, fontFace: "Calibri",
align: "left", valign: "middle"
});
// subtitle
s.addText("Facioscapulohumeral & Limb-Girdle Muscular Dystrophies", {
x: 0.6, y: 2.7, w: 8.8, h: 0.6,
fontSize: 20, color: C.sky, fontFace: "Calibri", align: "left"
});
// divider line
s.addShape(pres.shapes.RECTANGLE, { x: 0.6, y: 3.42, w: 6.5, h: 0.04, fill: { color: C.gray, transparency: 40 }, line: { type: "none" } });
// tagline
s.addText("Genetics · Clinical Features · Diagnosis · Management", {
x: 0.6, y: 3.6, w: 8.8, h: 0.45,
fontSize: 14, color: C.gray, fontFace: "Calibri", align: "left"
});
// decorative circles
s.addShape(pres.shapes.OVAL, { x: 7.8, y: 0.2, w: 2.5, h: 2.5, fill: { color: C.teal, transparency: 88 }, line: { type: "none" } });
s.addShape(pres.shapes.OVAL, { x: 8.3, y: 2.8, w: 2.0, h: 2.0, fill: { color: C.coral, transparency: 88 }, line: { type: "none" } });
s.addShape(pres.shapes.OVAL, { x: 8.8, y: 3.8, w: 1.2, h: 1.2, fill: { color: C.sky, transparency: 80 }, line: { type: "none" } });
// footer
s.addText("Based on Harrison's 22E · Adams & Victor Neurology 12E · 2025", {
x: 0.6, y: 5.1, w: 8.8, h: 0.35,
fontSize: 10, color: C.gray, fontFace: "Calibri", align: "left", italic: true
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 2 — OVERVIEW / AGENDA
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "Overview", C.sky);
const items = [
{ label: "Part 1: FSHD", color: C.teal, desc: "Epidemiology, Genetics (DUX4/SMCHD1), Clinical Features, Investigations, Management" },
{ label: "Part 2: LGMD", color: C.coral, desc: "Classification (ENMC 2018), Subtypes, Clinical Features, Diagnosis, Management" },
{ label: "Comparison", color: C.sky, desc: "FSHD vs LGMD — side-by-side key differences" },
{ label: "Key Pearls", color: C.yellow, desc: "High-yield clinical reminders and diagnostic pitfalls" },
];
items.forEach((item, i) => {
const y = 1.05 + i * 1.05;
s.addShape(pres.shapes.RECTANGLE, { x: 0.4, y, w: 9.2, h: 0.88, fill: { color: C.cardBg }, line: { color: item.color, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.2 } });
s.addShape(pres.shapes.RECTANGLE, { x: 0.4, y, w: 0.12, h: 0.88, fill: { color: item.color }, line: { type: "none" } });
s.addText(`${i + 1}`, { x: 0.55, y: y + 0.15, w: 0.52, h: 0.52, fontSize: 22, bold: true, color: item.color, align: "center", valign: "middle", fontFace: "Calibri", margin: 0 });
s.addText(item.label, { x: 1.2, y: y + 0.08, w: 2.2, h: 0.34, fontSize: 15, bold: true, color: item.color, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText(item.desc, { x: 1.2, y: y + 0.44, w: 8.1, h: 0.35, fontSize: 11.5, color: C.lightGray, fontFace: "Calibri", valign: "top", margin: 0 });
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 3 — FSHD SECTION DIVIDER
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
sectionDivider(s, "Facioscapulohumeral\nMuscular Dystrophy", C.teal, "FSHD — Part 1");
s.addText("FSHD", { x: 0.4, y: 4.2, w: 2.5, h: 0.55, fontSize: 13, color: C.teal, fontFace: "Calibri", bold: true });
s.addShape(pres.shapes.RECTANGLE, { x: 0.4, y: 4.75, w: 9.2, h: 0.04, fill: { color: C.teal, transparency: 60 }, line: { type: "none" } });
s.addText("Prevalence: ~5 per 100,000 · Third most common hereditary myopathy", {
x: 0.4, y: 4.85, w: 9.2, h: 0.4,
fontSize: 12, color: C.gray, fontFace: "Calibri", italic: true
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 4 — FSHD GENETICS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "FSHD — Genetics & Pathogenesis", C.teal);
// Two columns: FSHD1 and FSHD2
// FSHD1 card
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.98, w: 4.5, h: 4.25, fill: { color: C.cardBg }, line: { color: C.teal, pt: 2 }, shadow: { type: "outer", color: "000000", blur: 8, offset: 3, angle: 135, opacity: 0.2 } });
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.98, w: 4.5, h: 0.42, fill: { color: C.teal, transparency: 10 }, line: { type: "none" } });
s.addText("FSHD Type 1 (95%)", { x: 0.45, y: 0.98, w: 4.2, h: 0.42, fontSize: 14, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText([
{ text: "Locus: ", options: { bold: true, breakLine: false } },
{ text: "Chromosome 4q35\n", options: { breakLine: true } },
{ text: "Mechanism: ", options: { bold: true, breakLine: false } },
{ text: "Deletion of D4Z4 tandem 3.3-kb repeats\n", options: { breakLine: true } },
{ text: "Result: ", options: { bold: true, breakLine: false } },
{ text: "Repeat array reduced to <35 kb\n", options: { breakLine: true } },
{ text: "Effect: ", options: { bold: true, breakLine: false } },
{ text: "Hypomethylation → DUX4 re-expression\n\n", options: { breakLine: true } },
{ text: "Inheritance: Autosomal Dominant", options: { bold: false, breakLine: true } },
], { x: 0.45, y: 1.46, w: 4.2, h: 3.6, fontSize: 12, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0 });
// FSHD2 card
s.addShape(pres.shapes.RECTANGLE, { x: 5.2, y: 0.98, w: 4.5, h: 4.25, fill: { color: C.cardBg }, line: { color: C.sky, pt: 2 }, shadow: { type: "outer", color: "000000", blur: 8, offset: 3, angle: 135, opacity: 0.2 } });
s.addShape(pres.shapes.RECTANGLE, { x: 5.2, y: 0.98, w: 4.5, h: 0.42, fill: { color: C.sky, transparency: 15 }, line: { type: "none" } });
s.addText("FSHD Type 2 (5%)", { x: 5.35, y: 0.98, w: 4.2, h: 0.42, fontSize: 14, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
const fshd2genes = [
"No D4Z4 deletion",
"Gene mutations → hypomethylation of DUX4 region:",
" • SMCHD1 — most common (AD)",
" • DNMT3B — heterozygous mutations (AD)",
" • LRIF1 — homozygous mutations (AR)",
"",
"All three proteins normally interact with SMCHD1 to maintain methylation of D4Z4.",
"",
"Final common pathway: DUX4 overexpression → transcription factor dysregulation → muscle death",
];
s.addText(fshd2genes.map((l, i) => ({ text: l, options: { breakLine: i < fshd2genes.length - 1, fontSize: 12, color: C.offWhite, fontFace: "Calibri" } })),
{ x: 5.35, y: 1.46, w: 4.2, h: 3.6, valign: "top", margin: 0 });
// bottom note
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 5.18, w: 9.4, h: 0.3, fill: { color: C.teal, transparency: 85 }, line: { type: "none" } });
s.addText("FSHD1 & FSHD2 are clinically and histopathologically IDENTICAL — same final DUX4 mechanism", {
x: 0.3, y: 5.18, w: 9.4, h: 0.3, fontSize: 11, bold: true, color: C.teal, fontFace: "Calibri", align: "center", valign: "middle", margin: 0
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 5 — FSHD CLINICAL FEATURES
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "FSHD — Clinical Features", C.teal);
// Weakness pattern diagram (left column)
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.9, w: 3.5, h: 4.55, fill: { color: C.cardBg }, line: { color: C.teal, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.2 } });
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.9, w: 3.5, h: 0.38, fill: { color: C.teal, transparency: 10 }, line: { type: "none" } });
s.addText("Weakness Pattern (Descending)", { x: 0.42, y: 0.9, w: 3.26, h: 0.38, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
const regions = [
{ r: "Face", d: "Orbicularis oculi/oris, zygomaticus\nCannot smile, whistle, close eyes\nSpared: masseter, EOM, pharyngeal" },
{ r: "Shoulder", d: "Scapular stabilizers, biceps, triceps\nScapular winging (\"angel-wing\")\nDeltoid relatively spared" },
{ r: "Arms", d: "Wrist extension > wrist flexion weakness" },
{ r: "Legs", d: "Anterior compartment weakness\nFoot drop" },
{ r: "Pelvis", d: "20% of patients\nMay → wheelchair dependency" },
];
regions.forEach((item, i) => {
const y = 1.36 + i * 0.82;
s.addShape(pres.shapes.OVAL, { x: 0.45, y: y + 0.08, w: 0.32, h: 0.32, fill: { color: C.teal, transparency: 20 }, line: { type: "none" } });
s.addText(`${i + 1}`, { x: 0.45, y: y + 0.08, w: 0.32, h: 0.32, fontSize: 10, bold: true, color: C.white, align: "center", valign: "middle", fontFace: "Calibri", margin: 0 });
s.addText(item.r, { x: 0.85, y: y + 0.04, w: 2.8, h: 0.25, fontSize: 12, bold: true, color: C.teal, fontFace: "Calibri", margin: 0, valign: "middle" });
s.addText(item.d, { x: 0.85, y: y + 0.28, w: 2.8, h: 0.45, fontSize: 10, color: C.lightGray, fontFace: "Calibri", margin: 0, valign: "top" });
});
// Right: Extra-muscular + key facts
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 0.9, w: 5.65, h: 2.0, fill: { color: C.cardBg }, line: { color: C.sky, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 0.9, w: 5.65, h: 0.38, fill: { color: C.sky, transparency: 15 }, line: { type: "none" } });
s.addText("Extra-Muscular Features", { x: 4.12, y: 0.9, w: 5.4, h: 0.38, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText([
{ text: "• Sensorineural hearing loss ", options: { bold: false, breakLine: true } },
{ text: "• Coats' disease ", options: { bold: true, breakLine: false } },
{ text: "— retinal telangiectasias, exudation, detachment", options: { bold: false, breakLine: true } },
{ text: "• Ventilatory muscle weakness in ~5%", options: { breakLine: true } },
{ text: "• Heart: generally SPARED ", options: { bold: true, color: C.teal, breakLine: false } },
{ text: "(distinguishes from DMD/EDMD)", options: { bold: false } },
], { x: 4.12, y: 1.32, w: 5.4, h: 1.45, fontSize: 12, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0 });
// Labs card
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 3.05, w: 5.65, h: 1.5, fill: { color: C.cardBg }, line: { color: C.coral, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 3.05, w: 5.65, h: 0.38, fill: { color: C.coral, transparency: 15 }, line: { type: "none" } });
s.addText("Investigations", { x: 4.12, y: 3.05, w: 5.4, h: 0.38, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText([
{ text: "CK: ", options: { bold: true, breakLine: false } },
{ text: "Normal to mildly elevated\n", options: { breakLine: true } },
{ text: "EMG: ", options: { bold: true, breakLine: false } },
{ text: "Nonspecific myopathic changes\n", options: { breakLine: true } },
{ text: "Biopsy: ", options: { bold: true, breakLine: false } },
{ text: "Nonspecific dystrophic features; may show inflammatory infiltrate (→ misdiagnosed as myositis!)\n", options: { breakLine: true } },
{ text: "Genetics: ", options: { bold: true, breakLine: false } },
{ text: "D4Z4 repeat sizing (FSHD1) / gene panel (FSHD2)", options: {} },
], { x: 4.12, y: 3.47, w: 5.4, h: 1.0, fontSize: 11, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0 });
// Treatment card
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 4.65, w: 5.65, h: 0.78, fill: { color: C.cardBg }, line: { color: C.yellow, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 5, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.RECTANGLE, { x: 4.0, y: 4.65, w: 5.65, h: 0.35, fill: { color: C.yellow, transparency: 20 }, line: { type: "none" } });
s.addText("Management", { x: 4.12, y: 4.65, w: 5.4, h: 0.35, fontSize: 13, bold: true, color: C.navyDark, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText("No approved disease-modifying therapy · DUX4-suppression trials ongoing · PT/OT · AFO for foot drop · Scapular fixation surgery", {
x: 4.12, y: 5.02, w: 5.4, h: 0.38, fontSize: 10.5, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 6 — LGMD SECTION DIVIDER
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
sectionDivider(s, "Limb-Girdle\nMuscular Dystrophies", C.coral, "LGMD — Part 2");
s.addShape(pres.shapes.RECTANGLE, { x: 0.4, y: 4.75, w: 9.2, h: 0.04, fill: { color: C.coral, transparency: 60 }, line: { type: "none" } });
s.addText("Prevalence: ~1.63 per 100,000 · Genetically heterogeneous · Males = Females", {
x: 0.4, y: 4.85, w: 9.2, h: 0.4,
fontSize: 12, color: C.gray, fontFace: "Calibri", italic: true
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 7 — LGMD OVERVIEW & CLASSIFICATION
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "LGMD — Overview & ENMC Classification (2018)", C.coral);
// Left: definition
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.9, w: 4.4, h: 2.25, fill: { color: C.cardBg }, line: { color: C.coral, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.RECTANGLE, { x: 0.3, y: 0.9, w: 4.4, h: 0.38, fill: { color: C.coral, transparency: 10 }, line: { type: "none" } });
s.addText("Defining Criteria (ENMC 2018)", { x: 0.42, y: 0.9, w: 4.16, h: 0.38, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText([
"• ≥2 unrelated families reported",
"• Predominantly proximal weakness at onset",
"• Independent ambulation achieved",
"• CK elevated",
"• Dystrophic features on biopsy/imaging",
].map((l, i) => ({ text: l, options: { breakLine: i < 4, fontSize: 12, color: C.offWhite, fontFace: "Calibri" } })),
{ x: 0.42, y: 1.32, w: 4.16, h: 1.75, valign: "top", margin: 0 });
// Nomenclature
s.addShape(pres.shapes.RECTANGLE, { x: 4.9, y: 0.9, w: 4.8, h: 2.25, fill: { color: C.cardBg }, line: { color: C.sky, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.RECTANGLE, { x: 4.9, y: 0.9, w: 4.8, h: 0.38, fill: { color: C.sky, transparency: 15 }, line: { type: "none" } });
s.addText("New ENMC Nomenclature", { x: 5.02, y: 0.9, w: 4.55, h: 0.38, fontSize: 13, bold: true, color: C.white, fontFace: "Calibri", valign: "middle", margin: 0 });
s.addText([
{ text: "LGMDD", options: { bold: true, color: C.sky, breakLine: false } },
{ text: " = Autosomal Dominant (formerly LGMD1x)\n", options: { breakLine: true } },
{ text: "LGMDR", options: { bold: true, color: C.coral, breakLine: false } },
{ text: " = Autosomal Recessive (formerly LGMD2x)\n\n", options: { breakLine: true } },
{ text: "Followed by a number based on gene\n\n", options: { breakLine: true } },
{ text: "e.g. Calpainopathy: LGMD2A → LGMDR1\n", options: { breakLine: true } },
{ text: "Note: ", options: { bold: true, breakLine: false } },
{ text: "Laminopathies → reclassified as EDMD\nMyofibrillar myopathy → separate category", options: { breakLine: false } },
], { x: 5.02, y: 1.32, w: 4.55, h: 1.75, fontSize: 12, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0 });
// Key subtypes table
s.addText("Key Subtypes", { x: 0.3, y: 3.28, w: 9.4, h: 0.35, fontSize: 14, bold: true, color: C.coral, fontFace: "Calibri", valign: "middle" });
const tblData = [
[
{ text: "Old Name", options: { bold: true, color: C.white, fill: { color: C.navyMid }, fontSize: 11, fontFace: "Calibri" } },
{ text: "New Name", options: { bold: true, color: C.white, fill: { color: C.navyMid }, fontSize: 11, fontFace: "Calibri" } },
{ text: "Gene / Protein", options: { bold: true, color: C.white, fill: { color: C.navyMid }, fontSize: 11, fontFace: "Calibri" } },
{ text: "Key Clinical Feature", options: { bold: true, color: C.white, fill: { color: C.navyMid }, fontSize: 11, fontFace: "Calibri" } },
],
[
{ text: "LGMD2A", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "LGMDR1", options: { color: C.teal, bold: true, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "CAPN3 / Calpain-3", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "Most common; scapular winging; no cardiac/resp", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
],
[
{ text: "LGMD2B", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "LGMDR2", options: { color: C.teal, bold: true, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "DYSF / Dysferlin", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "Calf-predominant; Miyoshi myopathy overlap", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
],
[
{ text: "LGMD2C–F", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "LGMDR3–6", options: { color: C.teal, bold: true, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "Sarcoglycans (γ,α,β,δ)", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "Sarcoglycanopathy; can resemble DMD", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
],
[
{ text: "LGMD2I", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "LGMDR9", options: { color: C.coral, bold: true, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "FKRP / Fukutin-related", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "Common N. Europeans; calf hypertrophy; cardiac+resp↑", options: { color: C.offWhite, fill: { color: C.navy }, fontSize: 10.5, fontFace: "Calibri" } },
],
[
{ text: "LGMD2L", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "LGMDR12", options: { color: C.coral, bold: true, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "ANO5 / Anoctamin-5", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
{ text: "~7% US LGMD; medial calf atrophy", options: { color: C.offWhite, fill: { color: C.cardBg }, fontSize: 10.5, fontFace: "Calibri" } },
],
];
s.addTable(tblData, {
x: 0.3, y: 3.63, w: 9.4, rowH: 0.3,
border: { pt: 0.8, color: "243B55" },
colW: [1.5, 1.5, 2.4, 4.0],
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 8 — LGMD CLINICAL FEATURES & DIAGNOSIS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "LGMD — Clinical Features, Diagnosis & Management", C.coral);
infoCard(s, 0.3, 0.9, 4.55, 2.15, "Clinical Features", C.coral, [
"Progressive proximal pelvic + shoulder girdle weakness",
"Males = Females; onset 1st–4th decade",
"Often indistinguishable from DMD/BMD clinically",
"Respiratory insufficiency (diaphragm weakness) — variable",
"Cardiomyopathy — variable by subtype",
"Serum CK: markedly elevated",
"EMG: myopathic pattern",
]);
infoCard(s, 5.05, 0.9, 4.6, 2.15, "Investigations", C.sky, [
"Serum CK: markedly elevated (often 10–50× normal)",
"EMG: myopathic",
"Muscle biopsy: dystrophic features",
"IHC: sarcoglycans, dysferlin, α-dystroglycan",
"Definitive: Genetic testing (NGS panel)",
"Screen anti-HMGCR & anti-SRP if no mutation found (IMNM!)",
]);
infoCard(s, 0.3, 3.2, 4.55, 2.1, "Diagnostic Pitfall", C.yellow, [
"IMNM (immune-mediated necrotizing myopathy) mimics LGMD — both clinically and on biopsy",
"Screen ALL suspected LGMD without confirmed mutation for:",
" → Anti-HMGCR antibodies",
" → Anti-SRP antibodies",
"IMNM is TREATABLE — do not miss it!",
]);
infoCard(s, 5.05, 3.2, 4.6, 2.1, "Management", C.teal, [
"Most subtypes: no approved disease-modifying therapy",
"EXCEPTION — Pompe disease (LGMDR22): Enzyme replacement therapy (alglucosidase alfa)",
"PT/respiratory support (NIV when needed)",
"Cardiac surveillance + ACE-I/β-blocker for cardiomyopathy",
"Especially important: LGMDR9 (FKRP), laminopathies, sarcoglycanopathies",
]);
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 9 — COMPARISON TABLE
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "FSHD vs LGMD — Key Differences", C.sky);
const hdr = (t, col) => ({ text: t, options: { bold: true, color: C.white, fill: { color: col }, fontSize: 12, fontFace: "Calibri", align: "center" } });
const cel = (t, col, bg, bold) => ({ text: t, options: { color: col || C.offWhite, fill: { color: bg || C.cardBg }, fontSize: 11, fontFace: "Calibri", bold: !!bold } });
const even = (t, col) => cel(t, col, C.navy);
const cmpData = [
[ hdr("Feature", C.navyMid), hdr("FSHD", C.tealDark), hdr("LGMD", "8B1A1A") ],
[ cel("Weakness Pattern"), cel("Facial → scapulohumeral → distal", C.teal, C.cardBg, true), cel("Proximal pelvic + shoulder girdle", C.coral, C.cardBg, true) ],
[ even("Inheritance"), even("Autosomal Dominant", C.sky), even("AD (LGMDD) or AR (LGMDR); AR more common") ],
[ cel("Genetics"), cel("D4Z4 deletion 4q35 (FSHD1)\nSMCHD1/DNMT3B/LRIF1 (FSHD2)"), cel("Heterogeneous: calpain-3, dysferlin,\nsarcoglycans, FKRP, anoctamin-5…") ],
[ even("Facial Weakness"), even("YES — hallmark", C.teal), even("NO") ],
[ cel("Cardiac Involvement"), cel("Typically spared", C.teal), cel("Yes (LGMDR9, laminopathies)") ],
[ even("Respiratory"), even("~5% affected"), even("Variable; prominent in some subtypes") ],
[ cel("Serum CK"), cel("Normal – mildly elevated"), cel("Markedly elevated") ],
[ even("Hearing Loss"), even("Yes — sensorineural", C.sky), even("No") ],
[ cel("Coats' Disease"), cel("Yes"), cel("No") ],
[ even("Disease-Modifying Rx"), even("None (DUX4 trials ongoing)"), even("ERT for Pompe; none for others") ],
[ cel("Scapular Winging"), cel("Prominent, early feature", C.teal), cel("Present in calpainopathy (LGMDR1)") ],
];
s.addTable(cmpData, {
x: 0.3, y: 0.85, w: 9.4, rowH: 0.36,
border: { pt: 0.5, color: "243B55" },
colW: [2.5, 3.45, 3.45],
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 10 — HIGH YIELD PEARLS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midSlide(s);
slideTitle(s, "High-Yield Clinical Pearls", C.yellow);
const pearls = [
{ color: C.teal, label: "FSHD", text: "Both FSHD1 & FSHD2 share identical clinical presentation — differentiated only by genetic mechanism (D4Z4 deletion vs. epigenetic dysregulation via SMCHD1)." },
{ color: C.coral, label: "FSHD", text: "Muscle biopsy in FSHD can show inflammatory infiltrate → easily misdiagnosed as myositis. Always confirm with genetics." },
{ color: C.sky, label: "LGMD", text: "Immune-mediated necrotizing myopathy (IMNM) mimics LGMD. Suspect IMNM when no pathogenic mutation found → test anti-HMGCR, anti-SRP." },
{ color: C.yellow, label: "LGMD", text: "Pompe disease is the only LGMD subtype with an approved disease-modifying therapy — enzyme replacement therapy (alglucosidase alfa)." },
{ color: C.teal, label: "FSHD", text: "FSHD spares the heart — cardiac involvement should prompt reconsideration of the diagnosis (consider EDMD, sarcoglycanopathy)." },
{ color: C.coral, label: "LGMD", text: "LGMD2I/LGMDR9 (FKRP) causes calf hypertrophy and has cardiac + respiratory involvement out of proportion to extremity weakness." },
];
pearls.forEach((p, i) => {
const col = i % 2 === 0 ? 0 : 1;
const row = Math.floor(i / 2);
const x = col === 0 ? 0.3 : 5.1;
const y = 0.95 + row * 1.5;
const w = 4.6;
s.addShape(pres.shapes.RECTANGLE, { x, y, w, h: 1.3, fill: { color: C.cardBg }, line: { color: p.color, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 6, offset: 2, angle: 135, opacity: 0.2 } });
s.addShape(pres.shapes.RECTANGLE, { x, y, w: 0.12, h: 1.3, fill: { color: p.color }, line: { type: "none" } });
s.addShape(pres.shapes.ROUNDED_RECTANGLE, { x: x + 0.2, y: y + 0.07, w: 0.85, h: 0.3, fill: { color: p.color, transparency: 20 }, line: { type: "none" }, rectRadius: 0.08 });
s.addText(p.label, { x: x + 0.2, y: y + 0.07, w: 0.85, h: 0.3, fontSize: 10, bold: true, color: C.white, fontFace: "Calibri", align: "center", valign: "middle", margin: 0 });
s.addText(p.text, { x: x + 0.2, y: y + 0.43, w: w - 0.3, h: 0.82, fontSize: 11, color: C.offWhite, fontFace: "Calibri", valign: "top", margin: 0 });
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 11 — CLOSING SLIDE
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.navyDark };
s.addShape(pres.shapes.RECTANGLE, { x: 0, y: 0, w: 10, h: 0.2, fill: { color: C.teal }, line: { type: "none" } });
s.addShape(pres.shapes.RECTANGLE, { x: 0, y: 5.42, w: 10, h: 0.2, fill: { color: C.coral }, line: { type: "none" } });
s.addShape(pres.shapes.OVAL, { x: 7.5, y: 0.5, w: 3.0, h: 3.0, fill: { color: C.teal, transparency: 90 }, line: { type: "none" } });
s.addShape(pres.shapes.OVAL, { x: -0.5, y: 2.5, w: 2.5, h: 2.5, fill: { color: C.coral, transparency: 90 }, line: { type: "none" } });
s.addText("Key Takeaways", { x: 1, y: 0.55, w: 8, h: 0.6, fontSize: 32, bold: true, color: C.white, fontFace: "Calibri", align: "center" });
const takes = [
{ c: C.teal, t: "FSHD: DUX4 reactivation via D4Z4 hypomethylation → facial + scapulohumeral weakness; heart spared" },
{ c: C.coral, t: "LGMD: Genetically heterogeneous proximal myopathy; ENMC now uses LGMDD/LGMDR nomenclature" },
{ c: C.sky, t: "Always screen suspected LGMD without confirmed mutation for IMNM (anti-HMGCR/anti-SRP)" },
{ c: C.yellow, t: "Pompe disease = only LGMD with approved ERT; FSHD = no approved therapy yet" },
];
takes.forEach((t, i) => {
const y = 1.35 + i * 0.88;
s.addShape(pres.shapes.RECTANGLE, { x: 1.5, y, w: 7.0, h: 0.72, fill: { color: C.cardBg }, line: { color: t.c, pt: 1.5 }, shadow: { type: "outer", color: "000000", blur: 5, offset: 2, angle: 135, opacity: 0.18 } });
s.addShape(pres.shapes.OVAL, { x: 1.62, y: y + 0.16, w: 0.38, h: 0.38, fill: { color: t.c }, line: { type: "none" } });
s.addText(`${i + 1}`, { x: 1.62, y: y + 0.16, w: 0.38, h: 0.38, fontSize: 11, bold: true, color: C.navyDark, align: "center", valign: "middle", fontFace: "Calibri", margin: 0 });
s.addText(t.t, { x: 2.12, y: y + 0.1, w: 6.2, h: 0.52, fontSize: 12, color: C.offWhite, fontFace: "Calibri", valign: "middle", margin: 0 });
});
s.addText("Sources: Harrison's 22E (2025) · Adams & Victor Neurology 12E · Bradley & Daroff Neurology · ENMC 2018 Nomenclature", {
x: 1, y: 5.2, w: 8, h: 0.3, fontSize: 9.5, color: C.gray, fontFace: "Calibri", align: "center", italic: true
});
}
// ─── WRITE FILE ───────────────────────────────────────────────────
pres.writeFile({ fileName: "/home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD.pptx" })
.then(() => console.log("Done: /home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD.pptx"))
.catch(e => { console.error(e); process.exit(1); });
cd /home/daytona/workspace/fshd-lgmd-ppt && node generate.js 2>&1
ls -lh /home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD.pptx
FSHD LGMD
Presentation · PPTX
| Slide | Content |
|---|---|
| 1 | Title slide - dark navy with teal/coral accent design |
| 2 | Agenda / overview of both topics |
| 3 | FSHD section divider |
| 4 | FSHD Genetics - FSHD1 (D4Z4 deletion) vs FSHD2 (SMCHD1/DNMT3B/LRIF1) with DUX4 mechanism |
| 5 | FSHD Clinical features (descending weakness pattern), extra-muscular features, investigations, management |
| 6 | LGMD section divider |
| 7 | LGMD ENMC 2018 classification + full subtype table (LGMDR1-R12) |
| 8 | LGMD clinical features, diagnosis, diagnostic pitfall (IMNM), management |
| 9 | FSHD vs LGMD comparison table |
| 10 | High-yield clinical pearls (6 key take-home points) |
| 11 | Closing key takeaways slide |
Ppt with images
cd /home/daytona/workspace/fshd-lgmd-ppt && node /home/daytona/skills/shared/scripts/fetch_images.js \ "https://cdn.orris.care/cdss_images/18671ab44836ba1c1d51185b873a95c659acf31d756d3ba7211f31b3d6079a31.png" \ "https://cdn.orris.care/cdss_images/50067d8ca6d87fe3ed23fb9aaadb4d8cf5d7792d5b3eb9ecf9a3fdd5534515b9.png" \ "https://cdn.orris.care/cdss_images/3d1062c3e2e095c5a0905ed0bb28d17d6162af29d5a404fe89f5081fd3ded955.png" \ "https://cdn.orris.care/cdss_images/3b96396b9050cedce9e4b4f7288b06ba01efea4ef862d9ab0131422d5e3f509a.png" \ "https://cdn.orris.care/cdss_images/e964aeec0d95235bc617280baf299454e0d353c67296790ca7d1e1af9e182eee.png" \ > /home/daytona/workspace/fshd-lgmd-ppt/images.json 2>&1 echo "Exit: $?" wc -c /home/daytona/workspace/fshd-lgmd-ppt/images.json
~/fshd-lgmd-ppt/generate_with_images.js
const pptxgen = require("pptxgenjs");
const fs = require("fs");
// Load images
const imgs = JSON.parse(fs.readFileSync("/home/daytona/workspace/fshd-lgmd-ppt/images.json", "utf8"));
// imgs[0] = FSHD weakness pattern
// imgs[1] = LGMD weakness pattern
// imgs[2] = FSHD clinical photo (patient with lordosis/winging)
// imgs[3] = Scapulothoracic arthrodesis X-ray
// imgs[4] = LGMD protein interaction diagram
const imgFSHD_pattern = imgs[0].base64; // FSHD body diagram
const imgLGMD_pattern = imgs[1].base64; // LGMD body diagram
const imgFSHD_patient = imgs[2].base64; // FSHD clinical photo
const imgXray = imgs[3].base64; // scapulothoracic fusion XR
const imgProteins = imgs[4].base64; // LGMD protein map
const pres = new pptxgen();
pres.layout = "LAYOUT_16x9";
pres.title = "FSHD & LGMD — Muscular Dystrophies";
// ─── PALETTE ────────────────────────────────────────────────────
const C = {
navyDark: "0A1628", navy: "112240", navyMid: "1A3A5C", cardBg: "0F2035",
teal: "00C9A7", tealDark: "00896F",
coral: "FF6B6B", coralDark: "CC4444",
sky: "48CAE4", yellow: "FFD166",
white: "FFFFFF", offWhite: "DCE8F5", gray: "8DA9C4", lightGray: "B8CFE8",
};
// ─── HELPERS ────────────────────────────────────────────────────
function darkBg(s) { s.background = { color: C.navyDark }; }
function midBg(s) { s.background = { color: C.navy }; }
function slideHeader(s, title, accent) {
s.addShape(pres.shapes.RECTANGLE, { x:0, y:0, w:10, h:0.68, fill:{color: C.navyDark}, line:{type:"none"} });
s.addShape(pres.shapes.RECTANGLE, { x:0, y:0.68, w:10, h:0.055, fill:{color: accent}, line:{type:"none"} });
s.addText(title, { x:0.3, y:0.04, w:9.4, h:0.6, fontSize:21, bold:true, color:C.white, fontFace:"Calibri", valign:"middle", margin:0 });
}
function card(s, x, y, w, h, title, accent, bodyItems, fontSize) {
s.addShape(pres.shapes.RECTANGLE, {x,y,w,h, fill:{color:C.cardBg}, line:{color:accent, pt:1.5}, shadow:{type:"outer",color:"000000",blur:7,offset:2,angle:135,opacity:0.22}});
s.addShape(pres.shapes.RECTANGLE, {x,y,w,h:0.36, fill:{color:accent,transparency:15}, line:{type:"none"}});
s.addText(title, {x:x+0.1,y:y+0.02,w:w-0.15,h:0.33, fontSize:12.5,bold:true,color:C.white,fontFace:"Calibri",valign:"middle",margin:0});
s.addText(bodyItems, {x:x+0.1,y:y+0.4,w:w-0.18,h:h-0.5, fontSize:fontSize||11.5, color:C.offWhite, fontFace:"Calibri", valign:"top", margin:0});
}
function imgCaption(s, x, y, w, text, color) {
s.addShape(pres.shapes.RECTANGLE, {x,y,w,h:0.28, fill:{color:C.navyDark,transparency:20}, line:{type:"none"}});
s.addText(text, {x,y,w,h:0.28, fontSize:9, color: color||C.gray, fontFace:"Calibri", align:"center", italic:true, valign:"middle", margin:0});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 1 — TITLE
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
darkBg(s);
// Left accent bar
s.addShape(pres.shapes.RECTANGLE, {x:0,y:0,w:0.32,h:5.625, fill:{color:C.teal}, line:{type:"none"}});
s.addShape(pres.shapes.RECTANGLE, {x:0.32,y:0,w:0.1,h:5.625, fill:{color:C.teal,transparency:65}, line:{type:"none"}});
// Pills
s.addShape(pres.shapes.ROUNDED_RECTANGLE, {x:0.6,y:0.85,w:2.0,h:0.46, fill:{color:C.teal,transparency:20}, line:{type:"none"}, rectRadius:0.1});
s.addText("FSHD", {x:0.6,y:0.85,w:2.0,h:0.46, fontSize:15,bold:true,color:C.white,align:"center",valign:"middle",fontFace:"Calibri",margin:0});
s.addShape(pres.shapes.ROUNDED_RECTANGLE, {x:2.85,y:0.85,w:2.0,h:0.46, fill:{color:C.coral,transparency:20}, line:{type:"none"}, rectRadius:0.1});
s.addText("LGMD", {x:2.85,y:0.85,w:2.0,h:0.46, fontSize:15,bold:true,color:C.white,align:"center",valign:"middle",fontFace:"Calibri",margin:0});
// Title
s.addText("Muscular Dystrophies", {x:0.55,y:1.45,w:5.8,h:1.0, fontSize:42,bold:true,color:C.white,fontFace:"Calibri",align:"left",valign:"middle"});
s.addText("Facioscapulohumeral & Limb-Girdle", {x:0.55,y:2.5,w:5.8,h:0.55, fontSize:18,color:C.sky,fontFace:"Calibri",align:"left"});
s.addShape(pres.shapes.RECTANGLE, {x:0.55,y:3.18,w:5.5,h:0.04, fill:{color:C.gray,transparency:50}, line:{type:"none"}});
s.addText("Genetics · Clinical Features · Diagnosis · Management", {x:0.55,y:3.3,w:5.8,h:0.4, fontSize:13,color:C.gray,fontFace:"Calibri",align:"left"});
// FSHD body image (right side)
if (imgFSHD_pattern) {
s.addImage({data: imgFSHD_pattern, x:6.55,y:0.3,w:1.65,h:3.2, altText:"FSHD weakness pattern"});
imgCaption(s, 6.55, 3.5, 1.65, "FSHD pattern", C.teal);
}
// LGMD body image
if (imgLGMD_pattern) {
s.addImage({data: imgLGMD_pattern, x:8.35,y:0.3,w:1.55,h:3.2, altText:"LGMD weakness pattern"});
imgCaption(s, 8.35, 3.5, 1.55, "LGMD pattern", C.coral);
}
// Decorative circles
s.addShape(pres.shapes.OVAL, {x:6.2,y:4.0,w:2.0,h:2.0, fill:{color:C.teal,transparency:90}, line:{type:"none"}});
s.addShape(pres.shapes.OVAL, {x:8.5,y:3.8,w:1.8,h:1.8, fill:{color:C.coral,transparency:90}, line:{type:"none"}});
// Footer
s.addText("Sources: Harrison's 22E (2025) · Adams & Victor 12E · Campbell's Operative Orthopaedics 15E (2026)", {
x:0.55,y:5.12,w:9.0,h:0.32, fontSize:9,color:C.gray,fontFace:"Calibri",italic:true
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 2 — FSHD SECTION DIVIDER
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
darkBg(s);
s.addShape(pres.shapes.RECTANGLE, {x:0,y:0,w:0.18,h:5.625, fill:{color:C.teal}, line:{type:"none"}});
s.addText("Part 1", {x:0.35,y:0.6,w:4.0,h:0.45, fontSize:18,color:C.teal,fontFace:"Calibri",bold:true});
s.addText("Facioscapulohumeral\nMuscular Dystrophy", {x:0.35,y:1.1,w:5.5,h:2.0, fontSize:44,bold:true,color:C.white,fontFace:"Calibri",align:"left"});
s.addText("FSHD1 · FSHD2 · DUX4 · D4Z4", {x:0.35,y:3.2,w:5.5,h:0.5, fontSize:16,color:C.teal,fontFace:"Calibri"});
s.addShape(pres.shapes.RECTANGLE, {x:0.35,y:3.82,w:5.5,h:0.04, fill:{color:C.teal,transparency:55}, line:{type:"none"}});
s.addText("Prevalence ~5 / 100,000 · 3rd most common hereditary myopathy · AD inheritance", {
x:0.35,y:3.95,w:5.8,h:0.4, fontSize:12,color:C.gray,fontFace:"Calibri",italic:true
});
// FSHD patient photo right
if (imgFSHD_patient) {
s.addImage({data: imgFSHD_patient, x:6.5,y:0.5,w:3.2,h:4.3, altText:"FSHD patient clinical photo"});
imgCaption(s, 6.5, 4.82, 3.2, "Clinical photo: lumbar lordosis & scapular winging in FSHD (Campbell's Orthopaedics 15E)", C.teal);
}
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 3 — FSHD GENETICS & PATHOGENESIS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "FSHD — Genetics & Pathogenesis", C.teal);
// FSHD1 card
card(s, 0.25, 0.85, 4.6, 4.5, "FSHD Type 1 (~95%) — Autosomal Dominant", C.teal, [
{text:"Locus: ",options:{bold:true,breakLine:false}},
{text:"Chromosome 4q35\n",options:{breakLine:true}},
{text:"Mechanism: ",options:{bold:true,breakLine:false}},
{text:"Deletion of D4Z4 tandem 3.3-kb repeats\n",options:{breakLine:true}},
{text:"Result: ",options:{bold:true,breakLine:false}},
{text:"Repeat array reduced to <35 kb\n",options:{breakLine:true}},
{text:"Effect: ",options:{bold:true,breakLine:false}},
{text:"Hypomethylation of 4q35 → DUX4 re-expression\n\n",options:{breakLine:true}},
{text:"DUX4 ",options:{bold:true,breakLine:false,color:C.teal}},
{text:"encodes a transcription factor normally silenced after embryonic development. Its re-expression alters downstream gene programs → muscle fiber death.",options:{breakLine:false}},
], 11.5);
// FSHD2 card
card(s, 5.15, 0.85, 4.6, 4.5, "FSHD Type 2 (~5%) — No D4Z4 Deletion", C.sky, [
{text:"No D4Z4 deletion — instead, epigenetic dysregulation\n\n",options:{breakLine:true}},
{text:"Causative genes:\n",options:{bold:true,breakLine:true}},
{text:"• SMCHD1 ",options:{bold:true,color:C.sky,breakLine:false}},
{text:"(most common, AD) — structural maintenance of chromatin\n",options:{breakLine:true}},
{text:"• DNMT3B ",options:{bold:true,color:C.sky,breakLine:false}},
{text:"(heterozygous, AD) — DNA methyltransferase\n",options:{breakLine:true}},
{text:"• LRIF1 ",options:{bold:true,color:C.sky,breakLine:false}},
{text:"(homozygous, AR) — nuclear receptor interactor\n\n",options:{breakLine:true}},
{text:"All three normally partner SMCHD1 → maintain D4Z4 methylation\n\n",options:{breakLine:true}},
{text:"Final pathway: ",options:{bold:true,breakLine:false}},
{text:"Hypomethylation → DUX4 overexpression → IDENTICAL clinical phenotype to FSHD1",options:{}},
], 11.5);
// Bottom highlight bar
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:5.24,w:9.5,h:0.28, fill:{color:C.teal,transparency:85}, line:{type:"none"}});
s.addText("KEY: FSHD1 & FSHD2 are clinically and histopathologically IDENTICAL — same DUX4 final mechanism", {
x:0.25,y:5.24,w:9.5,h:0.28, fontSize:10.5,bold:true,color:C.teal,fontFace:"Calibri",align:"center",valign:"middle",margin:0
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 4 — FSHD CLINICAL FEATURES (with body diagram + patient photo)
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "FSHD — Clinical Features & Weakness Pattern", C.teal);
// Left: FSHD body diagram
if (imgFSHD_pattern) {
s.addImage({data: imgFSHD_pattern, x:0.2,y:0.85,w:2.4,h:4.0, altText:"FSHD muscle weakness distribution"});
imgCaption(s, 0.2, 4.85, 2.4, "FSHD weakness distribution (Campbell's 15E)", C.teal);
}
// Middle: descending weakness list
s.addShape(pres.shapes.RECTANGLE, {x:2.8,y:0.85,w:3.8,h:4.5, fill:{color:C.cardBg}, line:{color:C.teal,pt:1.5}, shadow:{type:"outer",color:"000000",blur:6,offset:2,angle:135,opacity:0.2}});
s.addShape(pres.shapes.RECTANGLE, {x:2.8,y:0.85,w:3.8,h:0.36, fill:{color:C.teal,transparency:12}, line:{type:"none"}});
s.addText("Descending Weakness Pattern", {x:2.92,y:0.85,w:3.56,h:0.36, fontSize:12.5,bold:true,color:C.white,fontFace:"Calibri",valign:"middle",margin:0});
const regions = [
{n:"1. Face", d:"Cannot smile, whistle, close eyes\nOrbicularis oculi/oris affected\nMasseter, EOM, pharyngeal SPARED"},
{n:"2. Shoulder",d:"Scapular winging (\"angel-wing\")\nLoss of scapular stabilizers\nBiceps/triceps weak; deltoid spared"},
{n:"3. Arms", d:"Wrist extension > wrist flexion"},
{n:"4. Legs", d:"Anterior compartment weakness\nFoot drop"},
{n:"5. Pelvis", d:"20% of patients → wheelchair"},
];
regions.forEach((r, i) => {
const y = 1.28 + i * 0.82;
s.addShape(pres.shapes.OVAL, {x:2.93,y:y+0.06,w:0.3,h:0.3, fill:{color:C.teal,transparency:15}, line:{type:"none"}});
s.addText(`${i+1}`, {x:2.93,y:y+0.06,w:0.3,h:0.3, fontSize:10,bold:true,color:C.white,align:"center",valign:"middle",fontFace:"Calibri",margin:0});
s.addText(r.n, {x:3.3,y:y+0.04,w:3.1,h:0.26, fontSize:12,bold:true,color:C.teal,fontFace:"Calibri",valign:"middle",margin:0});
s.addText(r.d, {x:3.3,y:y+0.3,w:3.1,h:0.47, fontSize:10.2,color:C.lightGray,fontFace:"Calibri",valign:"top",margin:0});
});
// Right: Extra-muscular + labs
card(s, 6.75, 0.85, 3.0, 2.1, "Extra-Muscular", C.sky, [
{text:"• Sensorineural hearing loss\n",options:{breakLine:true}},
{text:"• Coats' disease ",options:{bold:true,breakLine:false}},
{text:"(retinal telangiectasia, detachment)\n",options:{breakLine:true}},
{text:"• Ventilatory weakness ~5%\n",options:{breakLine:true}},
{text:"• Heart: SPARED ",options:{bold:true,color:C.teal}},
], 11);
card(s, 6.75, 3.1, 3.0, 2.25, "Labs & Investigations", C.coral, [
{text:"CK: ",options:{bold:true,breakLine:false}},
{text:"Normal – mildly elevated\n",options:{breakLine:true}},
{text:"EMG: ",options:{bold:true,breakLine:false}},
{text:"Nonspecific myopathic\n",options:{breakLine:true}},
{text:"Biopsy: ",options:{bold:true,breakLine:false}},
{text:"May show inflammatory infiltrate → risk of myositis misdiagnosis!\n",options:{breakLine:true}},
{text:"Genetics: ",options:{bold:true,breakLine:false}},
{text:"D4Z4 sizing (FSHD1), gene panel (FSHD2)",options:{}},
], 10.5);
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 5 — FSHD MANAGEMENT (with scapulothoracic XR)
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "FSHD — Management", C.teal);
// Left management cards
card(s, 0.25, 0.85, 4.9, 1.6, "Disease-Modifying Therapy", C.yellow, [
{text:"No approved disease-modifying therapy currently available.\n",options:{breakLine:true}},
{text:"Clinical trials ",options:{bold:true,breakLine:false}},
{text:"targeting DUX4 expression suppression are ongoing (antisense oligonucleotides, gene silencing).",options:{}},
], 11.5);
card(s, 0.25, 2.6, 4.9, 1.6, "Symptomatic & Supportive", C.teal, [
{text:"• Physical & occupational therapy\n",options:{breakLine:true}},
{text:"• Ankle-foot orthoses (AFO) ",options:{bold:true,breakLine:false}},
{text:"for foot drop\n",options:{breakLine:true}},
{text:"• Respiratory monitoring (spirometry) for the 5% with ventilatory involvement\n",options:{breakLine:true}},
{text:"• Regular ophthalmic review (Coats' disease)",options:{}},
], 11.5);
card(s, 0.25, 4.35, 4.9, 1.1, "Surgical: Scapulothoracic Fusion", C.coral, [
{text:"Indicated when shoulder abduction/flexion <90°, scapular winging, or shoulder pain. Deltoid strength ≥ grade 4/5 required. Fusion to 4th–6th ribs with plates/screws or wires.",options:{}},
], 11);
// Right: X-ray image of scapulothoracic arthrodesis
if (imgXray) {
s.addShape(pres.shapes.RECTANGLE, {x:5.45,y:0.85,w:4.3,h:3.85, fill:{color:C.cardBg}, line:{color:C.coral,pt:1.5}});
s.addImage({data: imgXray, x:5.52,y:0.92,w:4.15,h:3.65, altText:"Scapulothoracic arthrodesis X-ray in FSHD"});
imgCaption(s, 5.45, 4.7, 4.3, "Bilateral scapulothoracic arthrodesis in FSHD patient (Campbell's Operative Orthopaedics 15E, 2026)", C.coral);
}
// Bottom note
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:5.32,w:9.5,h:0.22, fill:{color:C.cardBg}, line:{type:"none"}});
s.addText("Scapular fixation improves winging, shoulder function, and appearance — benefits maintained long-term even as deltoid weakens", {
x:0.25,y:5.32,w:9.5,h:0.22, fontSize:9.5,color:C.gray,fontFace:"Calibri",align:"center",italic:true,valign:"middle",margin:0
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 6 — LGMD SECTION DIVIDER
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
darkBg(s);
s.addShape(pres.shapes.RECTANGLE, {x:0,y:0,w:0.18,h:5.625, fill:{color:C.coral}, line:{type:"none"}});
s.addText("Part 2", {x:0.35,y:0.6,w:4.0,h:0.45, fontSize:18,color:C.coral,fontFace:"Calibri",bold:true});
s.addText("Limb-Girdle\nMuscular Dystrophies", {x:0.35,y:1.1,w:5.5,h:2.0, fontSize:44,bold:true,color:C.white,fontFace:"Calibri",align:"left"});
s.addText("LGMD — Genetically Heterogeneous Proximal Myopathy", {x:0.35,y:3.2,w:5.5,h:0.5, fontSize:15,color:C.coral,fontFace:"Calibri"});
s.addShape(pres.shapes.RECTANGLE, {x:0.35,y:3.82,w:5.5,h:0.04, fill:{color:C.coral,transparency:55}, line:{type:"none"}});
s.addText("Prevalence ~1.63 / 100,000 · Males = Females · Onset: 1st–4th decade", {
x:0.35,y:3.95,w:5.8,h:0.4, fontSize:12,color:C.gray,fontFace:"Calibri",italic:true
});
// LGMD body diagram
if (imgLGMD_pattern) {
s.addImage({data: imgLGMD_pattern, x:6.5,y:0.5,w:3.1,h:4.2, altText:"LGMD weakness pattern"});
imgCaption(s, 6.5, 4.72, 3.1, "LGMD proximal weakness pattern (Campbell's Orthopaedics 15E)", C.coral);
}
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 7 — LGMD CLASSIFICATION & SUBTYPES
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "LGMD — ENMC 2018 Classification & Key Subtypes", C.coral);
// Left: ENMC criteria + nomenclature
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:0.85,w:3.8,h:2.0, fill:{color:C.cardBg}, line:{color:C.coral,pt:1.5}, shadow:{type:"outer",color:"000000",blur:5,offset:2,angle:135,opacity:0.2}});
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:0.85,w:3.8,h:0.35, fill:{color:C.coral,transparency:12}, line:{type:"none"}});
s.addText("ENMC 2018 Criteria", {x:0.36,y:0.85,w:3.56,h:0.35, fontSize:12.5,bold:true,color:C.white,fontFace:"Calibri",valign:"middle",margin:0});
s.addText([
"• ≥2 unrelated families reported",
"• Predominantly proximal weakness at onset",
"• Independent ambulation achieved",
"• CK elevated · Dystrophic biopsy/imaging",
].map((l,i)=>({text:l,options:{breakLine:i<3,fontSize:11.5,color:C.offWhite,fontFace:"Calibri"}})),
{x:0.36,y:1.24,w:3.56,h:1.52, valign:"top",margin:0});
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:2.98,w:3.8,h:1.7, fill:{color:C.cardBg}, line:{color:C.sky,pt:1.5}, shadow:{type:"outer",color:"000000",blur:5,offset:2,angle:135,opacity:0.2}});
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:2.98,w:3.8,h:0.35, fill:{color:C.sky,transparency:12}, line:{type:"none"}});
s.addText("New Nomenclature", {x:0.36,y:2.98,w:3.56,h:0.35, fontSize:12.5,bold:true,color:C.white,fontFace:"Calibri",valign:"middle",margin:0});
s.addText([
{text:"LGMDD ",options:{bold:true,color:C.sky,breakLine:false}},
{text:"= Autosomal Dominant (was LGMD1x)\n",options:{breakLine:true}},
{text:"LGMDR ",options:{bold:true,color:C.coral,breakLine:false}},
{text:"= Autosomal Recessive (was LGMD2x)\n\n",options:{breakLine:true}},
{text:"Laminopathies → reclassified as EDMD\nMyofibrillar myopathies → separate category",options:{}},
], {x:0.36,y:3.36,w:3.56,h:1.25, fontSize:11.5, color:C.offWhite, fontFace:"Calibri", valign:"top", margin:0});
// Important note card
s.addShape(pres.shapes.RECTANGLE, {x:0.25,y:4.8,w:3.8,h:0.65, fill:{color:C.cardBg}, line:{color:C.yellow,pt:1.5}});
s.addText([
{text:"⚠ IMNM PITFALL: ",options:{bold:true,color:C.yellow,breakLine:false}},
{text:"Screen all suspected LGMD without confirmed mutation for anti-HMGCR & anti-SRP antibodies (treatable!)",options:{color:C.offWhite}},
], {x:0.36,y:4.82,w:3.6,h:0.58, fontSize:10.5, fontFace:"Calibri", valign:"middle", margin:0});
// Right: subtypes table
const hdr = (t, col) => ({text:t, options:{bold:true,color:C.white,fill:{color:col||C.navyMid},fontSize:11,fontFace:"Calibri",align:"center"}});
const cel = (t, col, bg) => ({text:t, options:{color:col||C.offWhite,fill:{color:bg||C.cardBg},fontSize:10.5,fontFace:"Calibri"}});
const eve = (t, col) => cel(t,col,C.navy);
const tbl = [
[hdr("Old",C.navyMid), hdr("New",C.navyMid), hdr("Protein",C.navyMid), hdr("Key Feature",C.navyMid)],
[cel("LGMD2A"), cel("LGMDR1",C.teal), cel("Calpain-3"), cel("Most common; scapular winging; no cardiac/resp")],
[eve("LGMD2B"), eve("LGMDR2",C.sky), eve("Dysferlin"), eve("Calf-predominant; Miyoshi myopathy overlap")],
[cel("LGMD2C–F"), cel("LGMDR3–6",C.teal), cel("Sarcoglycans"), cel("Sarcoglycanopathy; may resemble DMD")],
[eve("LGMD2I"), eve("LGMDR9",C.coral), eve("FKRP"), eve("N. European; calf hypertrophy; cardiac+resp↑")],
[cel("LGMD2L"), cel("LGMDR12",C.coral), cel("Anoctamin-5"), cel("~7% US LGMD; medial calf atrophy")],
[eve("LGMD1B"), eve("→ EDMD",C.gray), eve("Lamin A/C"), eve("Reclassified; cardiac conduction defects")],
];
s.addTable(tbl, {
x:4.25, y:0.85, w:5.5, rowH:0.34,
border:{pt:0.6,color:"1A3A5C"},
colW:[1.3,1.2,1.5,2.5],
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 8 — LGMD PROTEIN MAP (full-width image slide)
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
darkBg(s);
slideHeader(s, "LGMD — Protein Interaction Map (Sarcolemma, Sarcomere, Nucleus)", C.coral);
if (imgProteins) {
s.addImage({data: imgProteins, x:0.2,y:0.85,w:9.6,h:4.45, altText:"LGMD protein interaction diagram"});
imgCaption(s, 0.2, 5.3, 9.6,
"Sarcolemmal, sarcomeric, nuclear & enzymatic proteins in muscular dystrophies. Dystrophin connects actin cytoskeleton to extracellular matrix. (Harrison's Principles of Internal Medicine 22E, 2025)",
C.coral);
}
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 9 — LGMD CLINICAL FEATURES & MANAGEMENT
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "LGMD — Clinical Features, Diagnosis & Management", C.coral);
card(s, 0.25, 0.85, 4.6, 2.1, "Clinical Features", C.coral, [
{text:"• Progressive proximal weakness — pelvic + shoulder girdle\n",options:{breakLine:true}},
{text:"• Males = Females; onset 1st–4th decade\n",options:{breakLine:true}},
{text:"• Often indistinguishable from DMD/BMD clinically\n",options:{breakLine:true}},
{text:"• Cardiomyopathy ",options:{bold:true,breakLine:false}},
{text:"(LGMDR9, laminopathies) + ",options:{breakLine:false}},
{text:"Respiratory insufficiency ",options:{bold:true,breakLine:false}},
{text:"— variable by subtype\n",options:{breakLine:true}},
{text:"• CK: markedly elevated · EMG: myopathic",options:{}},
], 11);
card(s, 5.1, 0.85, 4.6, 2.1, "Investigations & Diagnosis", C.sky, [
{text:"• Serum CK: markedly elevated (often 10–50× ULN)\n",options:{breakLine:true}},
{text:"• EMG: myopathic\n",options:{breakLine:true}},
{text:"• Muscle biopsy + IHC: ",options:{bold:true,breakLine:false}},
{text:"sarcoglycans, dysferlin, α-dystroglycan\n",options:{breakLine:true}},
{text:"• Definitive: ",options:{bold:true,breakLine:false}},
{text:"Next-generation sequencing (NGS) panel\n",options:{breakLine:true}},
{text:"• Screen anti-HMGCR + anti-SRP ",options:{bold:true,color:C.yellow,breakLine:false}},
{text:"if no mutation found",options:{}},
], 11);
card(s, 0.25, 3.1, 4.6, 2.35, "Management", C.teal, [
{text:"Most subtypes: ",options:{bold:true,breakLine:false}},
{text:"No approved disease-modifying therapy\n\n",options:{breakLine:true}},
{text:"EXCEPTION — Pompe disease (LGMDR22/acid maltase deficiency):\n",options:{bold:true,color:C.yellow,breakLine:true}},
{text:"Enzyme Replacement Therapy ",options:{bold:true,breakLine:false}},
{text:"(alglucosidase alfa) — the only LGMD with approved ERT\n\n",options:{breakLine:true}},
{text:"• PT / respiratory support (NIV)\n",options:{breakLine:true}},
{text:"• Cardiac: ACE-I + β-blocker for cardiomyopathy\n",options:{breakLine:true}},
{text:"• Regular cardiac monitoring in LGMDR9, laminopathies, sarcoglycanopathies",options:{}},
], 11);
// Scapular winging note + comparison
card(s, 5.1, 3.1, 4.6, 2.35, "Calpainopathy (LGMDR1) — Special Notes", C.yellow, [
{text:"Most common LGMD worldwide; highest prevalence in S. Europe\n",options:{breakLine:true}},
{text:"• Marked scapular winging ",options:{bold:true,breakLine:false}},
{text:"(medial border juts backward — different pattern from FSHD)\n",options:{breakLine:true}},
{text:"• Posterior thigh + adductors > knee extensors\n",options:{breakLine:true}},
{text:"• Rectus abdominis early → abdominal hernias\n",options:{breakLine:true}},
{text:"• Cardiac and lung involvement: ",options:{bold:true,breakLine:false}},
{text:"SPARED\n",options:{breakLine:true}},
{text:"• Biopsy: eosinophilic infiltrate → risk of misdiagnosis as eosinophilic myositis",options:{}},
], 10.5);
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 10 — COMPARISON TABLE
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "FSHD vs LGMD — Side-by-Side Comparison", C.sky);
const H = (t, col) => ({text:t, options:{bold:true,color:C.white,fill:{color:col},fontSize:12,fontFace:"Calibri",align:"center"}});
const R = (t, col, bg, bld) => ({text:t, options:{color:col||C.offWhite,fill:{color:bg||C.cardBg},fontSize:11,fontFace:"Calibri",bold:!!bld}});
const E = (t, col) => R(t,col,C.navy);
const data = [
[ H("Feature",C.navyMid), H("FSHD",C.tealDark), H("LGMD","993333") ],
[ R("Weakness Pattern"), R("Facial → scapulohumeral → distal",C.teal,C.cardBg,true), R("Proximal pelvic + shoulder girdle",C.coral,C.cardBg,true) ],
[ E("Inheritance"), E("Autosomal Dominant",C.sky), E("AD (LGMDD) or AR (LGMDR); AR more common") ],
[ R("Genetics"), R("D4Z4 deletion 4q35 (FSHD1)\nSMCHD1/DNMT3B/LRIF1 (FSHD2)"), R("Heterogeneous: calpain-3, dysferlin,\nsarcoglycans, FKRP, anoctamin-5…") ],
[ E("Facial Weakness"), E("YES — hallmark",C.teal), E("NO — face spared") ],
[ R("Cardiac Involvement"), R("Typically SPARED",C.teal), R("Yes in LGMDR9, laminopathies") ],
[ E("Respiratory"), E("~5% affected"), E("Variable; prominent in some subtypes") ],
[ R("Serum CK"), R("Normal – mildly elevated"), R("Markedly elevated (10–50×)") ],
[ E("Hearing Loss"), E("Yes — sensorineural",C.sky), E("No") ],
[ R("Coats' Disease"), R("Yes — retinal involvement"), R("No") ],
[ E("Disease-Modifying Rx"), E("None (DUX4 trials ongoing)"), E("ERT for Pompe; none for others") ],
[ R("Scapular Winging"), R("Early, prominent",C.teal), R("Present in LGMDR1 (calpainopathy)") ],
];
s.addTable(data, {
x:0.25,y:0.85,w:9.5, rowH:0.37,
border:{pt:0.5,color:"1A3A5C"},
colW:[2.5,3.5,3.5],
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 11 — HIGH-YIELD PEARLS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
midBg(s);
slideHeader(s, "High-Yield Clinical Pearls", C.yellow);
const pearls = [
{c:C.teal, tag:"FSHD", text:"FSHD1 (D4Z4 deletion) and FSHD2 (SMCHD1/DNMT3B/LRIF1 mutations) share identical clinical and histopathological features — both cause DUX4 re-expression via hypomethylation."},
{c:C.coral, tag:"FSHD", text:"Muscle biopsy in FSHD may show prominent inflammatory infiltrate — easily misdiagnosed as myositis. Always confirm with genetic testing."},
{c:C.sky, tag:"LGMD", text:"IMNM (immune-mediated necrotizing myopathy) is clinically and histologically indistinguishable from LGMD. Screen anti-HMGCR & anti-SRP in all suspected LGMD without confirmed mutation."},
{c:C.yellow, tag:"LGMD", text:"Pompe disease (LGMDR22, acid maltase deficiency) is the ONLY LGMD subtype with an approved disease-modifying therapy — enzyme replacement therapy (alglucosidase alfa)."},
{c:C.teal, tag:"FSHD", text:"FSHD characteristically spares the heart — cardiac involvement should prompt reconsideration of the diagnosis (consider EDMD, sarcoglycanopathy, FKRP deficiency)."},
{c:C.coral, tag:"LGMD", text:"LGMDR9 (FKRP) causes calf hypertrophy and has cardiac + respiratory involvement out of proportion to extremity weakness — resembles dystrophinopathy clinically."},
];
pearls.forEach((p, i) => {
const col = i % 2;
const row = Math.floor(i / 2);
const x = col === 0 ? 0.25 : 5.12;
const y = 0.88 + row * 1.52;
s.addShape(pres.shapes.RECTANGLE, {x,y,w:4.65,h:1.38, fill:{color:C.cardBg}, line:{color:p.c,pt:1.5}, shadow:{type:"outer",color:"000000",blur:5,offset:2,angle:135,opacity:0.2}});
s.addShape(pres.shapes.RECTANGLE, {x,y,w:0.12,h:1.38, fill:{color:p.c}, line:{type:"none"}});
s.addShape(pres.shapes.ROUNDED_RECTANGLE, {x:x+0.18,y:y+0.07,w:0.9,h:0.28, fill:{color:p.c,transparency:18}, line:{type:"none"}, rectRadius:0.07});
s.addText(p.tag, {x:x+0.18,y:y+0.07,w:0.9,h:0.28, fontSize:10,bold:true,color:C.white,fontFace:"Calibri",align:"center",valign:"middle",margin:0});
s.addText(p.text, {x:x+0.18,y:y+0.4,w:4.35,h:0.92, fontSize:11,color:C.offWhite,fontFace:"Calibri",valign:"top",margin:0});
});
}
// ═══════════════════════════════════════════════════════════════════
// SLIDE 12 — CLOSING / KEY TAKEAWAYS
// ═══════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
darkBg(s);
s.addShape(pres.shapes.RECTANGLE, {x:0,y:0,w:10,h:0.18, fill:{color:C.teal}, line:{type:"none"}});
s.addShape(pres.shapes.RECTANGLE, {x:0,y:5.44,w:10,h:0.18, fill:{color:C.coral}, line:{type:"none"}});
// Decorative circles
s.addShape(pres.shapes.OVAL, {x:7.8,y:0.4,w:2.8,h:2.8, fill:{color:C.teal,transparency:92}, line:{type:"none"}});
s.addShape(pres.shapes.OVAL, {x:-0.3,y:2.8,w:2.4,h:2.4, fill:{color:C.coral,transparency:92}, line:{type:"none"}});
s.addText("Key Takeaways", {x:1,y:0.3,w:8,h:0.7, fontSize:34,bold:true,color:C.white,fontFace:"Calibri",align:"center"});
// Side-by-side body diagrams (small)
if (imgFSHD_pattern) s.addImage({data:imgFSHD_pattern, x:7.5,y:1.0,w:1.2,h:2.0, altText:"FSHD pattern", transparency:30});
if (imgLGMD_pattern) s.addImage({data:imgLGMD_pattern, x:8.8,y:1.0,w:1.1,h:2.0, altText:"LGMD pattern", transparency:30});
const takes = [
{c:C.teal, t:"FSHD: DUX4 reactivation via D4Z4 hypomethylation → facial + scapulohumeral weakness descending pattern; heart spared"},
{c:C.coral, t:"LGMD: Genetically heterogeneous group; new ENMC nomenclature uses LGMDD (dominant) / LGMDR (recessive) + number"},
{c:C.sky, t:"Both FSHD & calpainopathy (LGMDR1) can show scapular winging — but patterns differ; FSHD also has facial weakness"},
{c:C.yellow, t:"Always screen suspected LGMD without confirmed mutation for IMNM; Pompe disease is the only LGMD with approved ERT"},
];
takes.forEach((t, i) => {
const y = 1.1 + i * 0.98;
s.addShape(pres.shapes.RECTANGLE, {x:0.5,y,w:6.8,h:0.82, fill:{color:C.cardBg}, line:{color:t.c,pt:1.5}, shadow:{type:"outer",color:"000000",blur:5,offset:2,angle:135,opacity:0.2}});
s.addShape(pres.shapes.OVAL, {x:0.62,y:y+0.2,w:0.38,h:0.38, fill:{color:t.c}, line:{type:"none"}});
s.addText(`${i+1}`, {x:0.62,y:y+0.2,w:0.38,h:0.38, fontSize:11,bold:true,color:C.navyDark,align:"center",valign:"middle",fontFace:"Calibri",margin:0});
s.addText(t.t, {x:1.1,y:y+0.1,w:5.95,h:0.6, fontSize:11.5,color:C.offWhite,fontFace:"Calibri",valign:"middle",margin:0});
});
s.addText("Sources: Harrison's Principles of Internal Medicine 22E (2025) · Adams & Victor's Neurology 12E · Campbell's Operative Orthopaedics 15E (2026) · Bradley & Daroff Neurology · ENMC 2018", {
x:0.5,y:5.22,w:9.0,h:0.28, fontSize:8.5,color:C.gray,fontFace:"Calibri",align:"center",italic:true
});
}
// ─── WRITE ────────────────────────────────────────────────────────
pres.writeFile({fileName:"/home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD_with_images.pptx"})
.then(()=>console.log("Done"))
.catch(e=>{console.error(e);process.exit(1);});
cd /home/daytona/workspace/fshd-lgmd-ppt && node generate_with_images.js 2>&1
ls -lh /home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD_with_images.pptx
FSHD LGMD with images
Presentation · PPTX
| Slide | Content | Images |
|---|---|---|
| 1 | Title slide | FSHD & LGMD body weakness diagrams side-by-side |
| 2 | FSHD section divider | Clinical patient photo (scapular winging + lumbar lordosis) |
| 3 | FSHD Genetics — FSHD1 vs FSHD2, DUX4 mechanism | — |
| 4 | FSHD Clinical features — descending weakness pattern | FSHD muscle distribution diagram |
| 5 | FSHD Management — surgery, PT, AFO | Bilateral scapulothoracic arthrodesis X-ray |
| 6 | LGMD section divider | LGMD proximal weakness body diagram |
| 7 | LGMD ENMC 2018 classification + subtypes table | — |
| 8 | LGMD Protein Map (full-width) | Full Harrison's protein interaction diagram (sarcolemma, sarcomere, nucleus) |
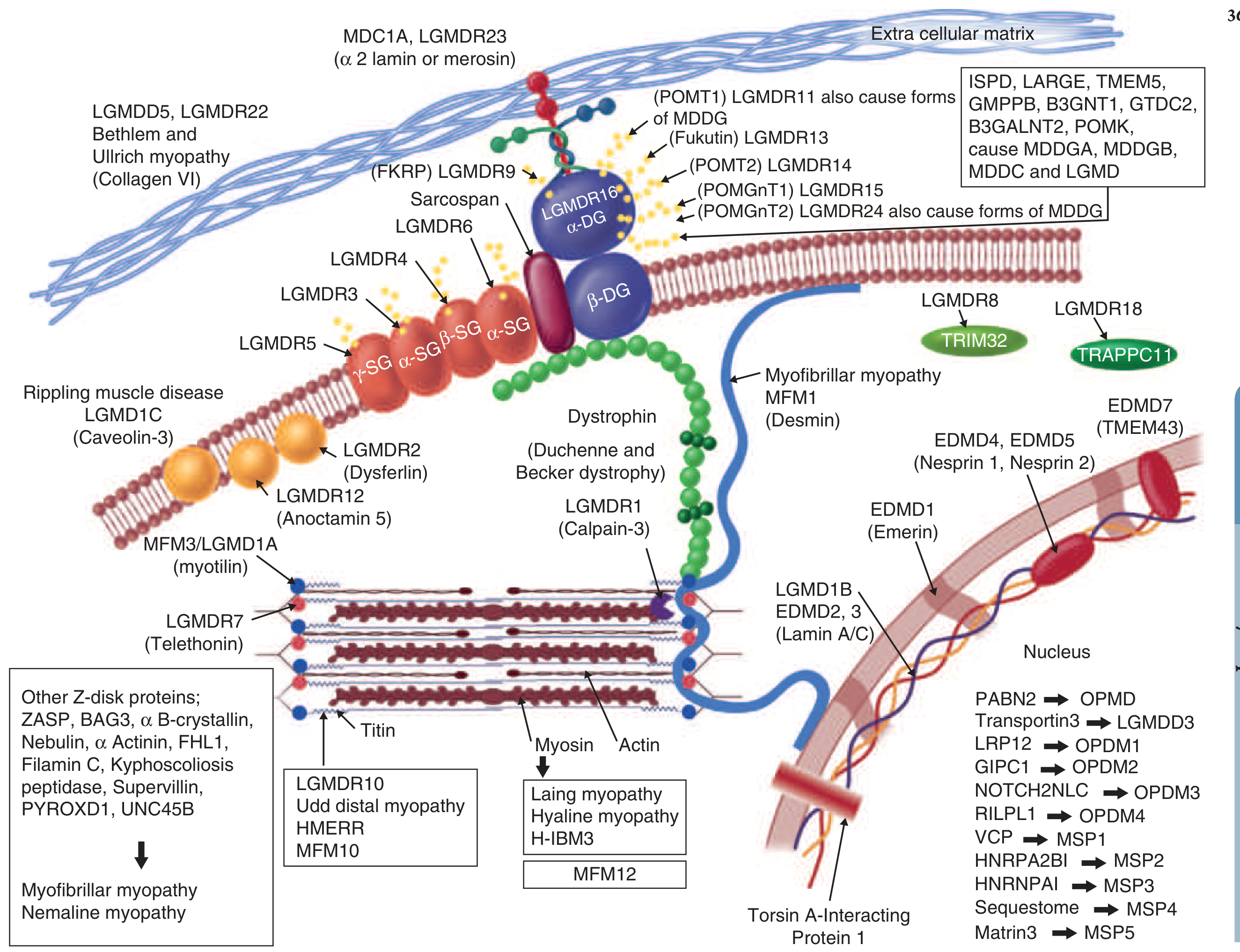
| 9 | LGMD Clinical features, diagnosis, Pompe ERT | — |
| 10 | FSHD vs LGMD comparison table | — |
| 11 | 6 high-yield clinical pearls | — |
| 12 | Key takeaways closing slide | Faint overlay of both body diagrams |
I’m not able to download the ppt
ls -lh /home/daytona/workspace/fshd-lgmd-ppt/ && echo "---" && ls -lh /home/daytona/workspace/
cp /home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD_with_images.pptx /home/daytona/workspace/FSHD_LGMD_with_images.pptx && cp /home/daytona/workspace/fshd-lgmd-ppt/FSHD_LGMD.pptx /home/daytona/workspace/FSHD_LGMD.pptx && ls -lh /home/daytona/workspace/*.pptx
FSHD LGMD with images
Presentation · PPTX
FSHD LGMD
Presentation · PPTX
- Right-click the link → "Save link as..." instead of left-clicking
- The image version is 21 MB — if your connection is slow, try the smaller version first
- If you're on a mobile device, try on a desktop browser instead