Pancreatic enzymes and there function
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive overview of pancreatic enzymes and their functions, drawn from multiple authoritative sources.
Pancreatic Enzymes and Their Functions
The exocrine pancreas secretes a juice containing enzymes for digesting all three major macronutrients — proteins, carbohydrates, and fats — along with bicarbonate (HCO₃⁻) to neutralize gastric acid entering the duodenum. — Guyton and Hall Textbook of Medical Physiology
1. Proteolytic Enzymes (Protein Digestion)
All pancreatic proteases are secreted as inactive zymogens to prevent autodigestion of the pancreas.
| Zymogen | Active Enzyme | Function |
|---|---|---|
| Trypsinogen | Trypsin | Cleaves proteins/peptides at basic AA (Arg, Lys); also activates all other zymogens |
| Chymotrypsinogen | Chymotrypsin | Cleaves at aromatic/bulky AA (Phe, Trp, Tyr) |
| Proelastase | Elastase | Cleaves at small neutral AA (Ala, Gly, Val); digests elastin |
| Procarboxypeptidase A | Carboxypeptidase A | Exopeptidase; removes aromatic/aliphatic AA from the C-terminus |
| Procarboxypeptidase B | Carboxypeptidase B | Exopeptidase; removes basic AA (Arg, Lys) from the C-terminus |
Activation cascade: The brush-border enzyme enterokinase (enteropeptidase) converts trypsinogen → trypsin. Trypsin then autocatalyzes itself and activates all other zymogens. — Costanzo Physiology, 7th ed.
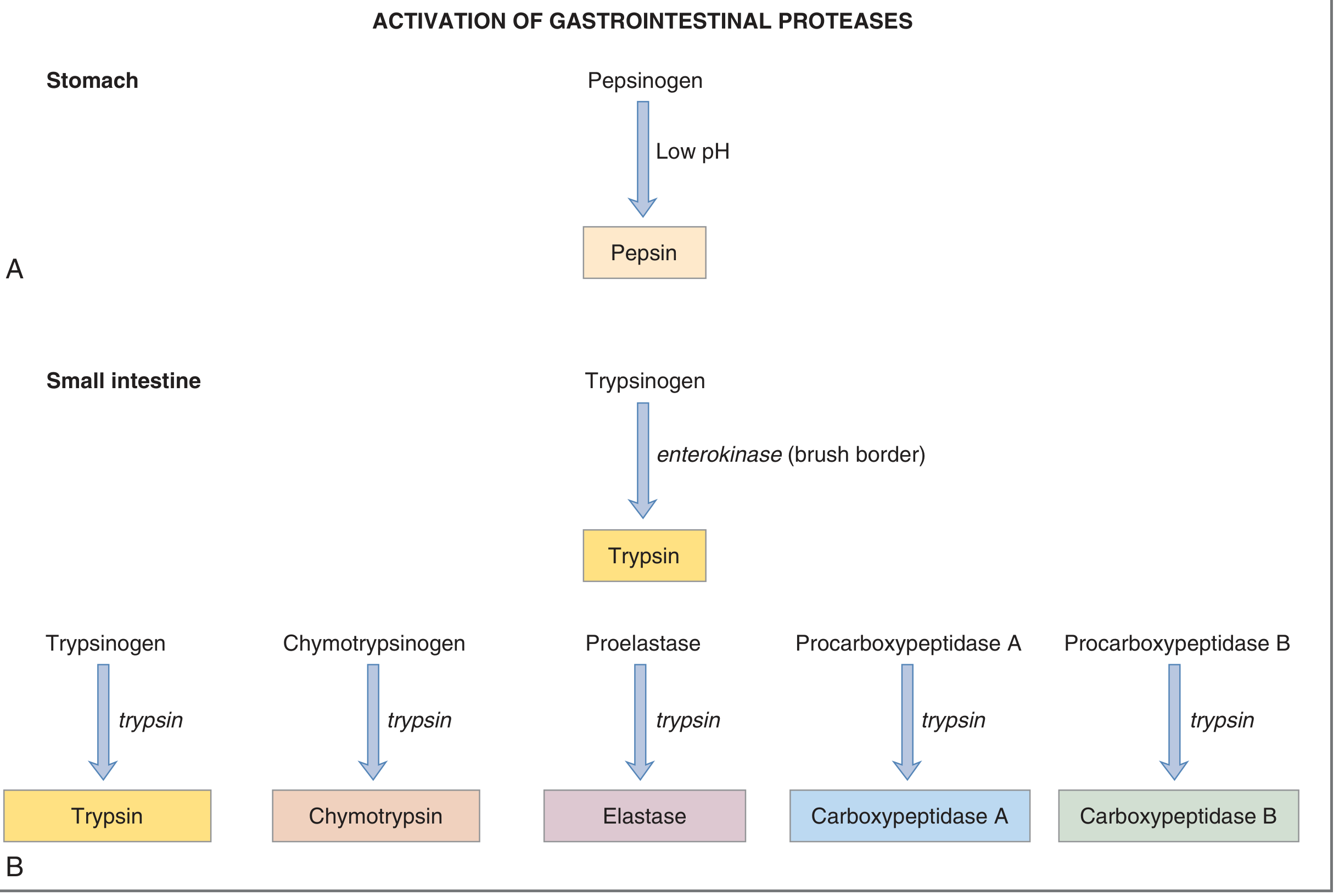
The products of proteolysis are amino acids, dipeptides, tripeptides, and oligopeptides. Only amino acids, dipeptides, and tripeptides are directly absorbable; oligopeptides are further hydrolyzed by brush-border proteases.
2. Amylolytic Enzyme (Carbohydrate Digestion)
| Enzyme | Function |
|---|---|
| Pancreatic amylase | Hydrolyzes starches, glycogen, and most polysaccharides (except cellulose) → disaccharides and trisaccharides (mainly maltose) |
Pancreatic amylase does not digest cellulose (humans lack the necessary enzyme). Final digestion to monosaccharides is completed by brush-border enzymes (maltase, sucrase, lactase). — Guyton and Hall
3. Lipolytic Enzymes (Fat Digestion)
| Enzyme | Zymogen | Function |
|---|---|---|
| Pancreatic lipase | — (active as secreted) | Hydrolyzes triacylglycerols (TAG) at C-1 and C-3 positions → 2-monoacylglycerol + 2 free fatty acids |
| Colipase | Procolipase | Not an enzyme itself; co-secreted with lipase, anchors it to the lipid-aqueous interface, restoring its activity in the presence of inhibitory bile salts |
| Cholesterol esterase (cholesterol ester hydrolase) | — | Hydrolyzes cholesterol esters → free cholesterol + fatty acids; activity enhanced by bile salts |
| Phospholipase A2 | Pro-phospholipase A2 | Removes the fatty acid at C-2 of phospholipids → lysophospholipid + free fatty acid; activated by trypsin, requires bile salts |
Pancreatic lipase constitutes 2–3% of total protein in pancreatic secretions and is highly efficient — significant fat malabsorption only occurs in severe pancreatic insufficiency (e.g., cystic fibrosis). — Lippincott's Illustrated Reviews: Biochemistry, 8th ed.
4. Protective Mechanism: Trypsin Inhibitor
The same acinar cells that secrete proteolytic zymogens also secrete pancreatic secretory trypsin inhibitor (PSTI). This inhibitor blocks premature trypsin activation within the pancreatic ducts. If this system is overwhelmed (e.g., duct obstruction, trauma), activated enzymes digest pancreatic tissue itself — producing acute pancreatitis. — Guyton and Hall
5. Hormonal Regulation of Secretion
| Hormone | Source | Stimulus | Effect on Pancreas |
|---|---|---|---|
| Cholecystokinin (CCK) | I cells of duodenum/jejunum | Fats and proteins in duodenum | Stimulates secretion of digestive enzymes |
| Secretin | S cells of duodenum | Low pH of chyme | Stimulates secretion of bicarbonate-rich fluid |
CCK also slows gastric emptying to allow adequate digestion time, while secretin's bicarbonate neutralizes gastric acid and brings the intestinal pH to the optimal range for pancreatic enzyme activity. — Lippincott's Illustrated Reviews: Biochemistry, 8th ed.
Summary Table
| Enzyme | Substrate | Product |
|---|---|---|
| Trypsin | Proteins/peptides | Smaller peptides |
| Chymotrypsin | Proteins/peptides | Smaller peptides |
| Elastase | Proteins/elastin | Smaller peptides |
| Carboxypeptidase A & B | Peptides | Amino acids (from C-terminus) |
| Pancreatic amylase | Starch, glycogen | Disaccharides, trisaccharides |
| Pancreatic lipase | Triglycerides | 2-MAG + fatty acids |
| Cholesterol esterase | Cholesterol esters | Cholesterol + fatty acids |
| Phospholipase A2 | Phospholipids | Lysophospholipid + fatty acid |
Gilbert syndrome and types of jaundice
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Excellent — I now have comprehensive, authoritative content from multiple textbooks. Here is the full answer.
Types of Jaundice
Jaundice is yellow discoloration of the skin and sclerae caused by excess bilirubin (hyperbilirubinemia) in the plasma. It becomes clinically visible when serum bilirubin exceeds ~2–3 mg/dL. — Gray's Anatomy for Students
Hyperbilirubinemia arises from: (1) overproduction of bilirubin; (2) impaired uptake, conjugation, or excretion; or (3) regurgitation from damaged hepatocytes or bile ducts. — Harrison's Principles of Internal Medicine, 22nd ed.
The Three Anatomical Types
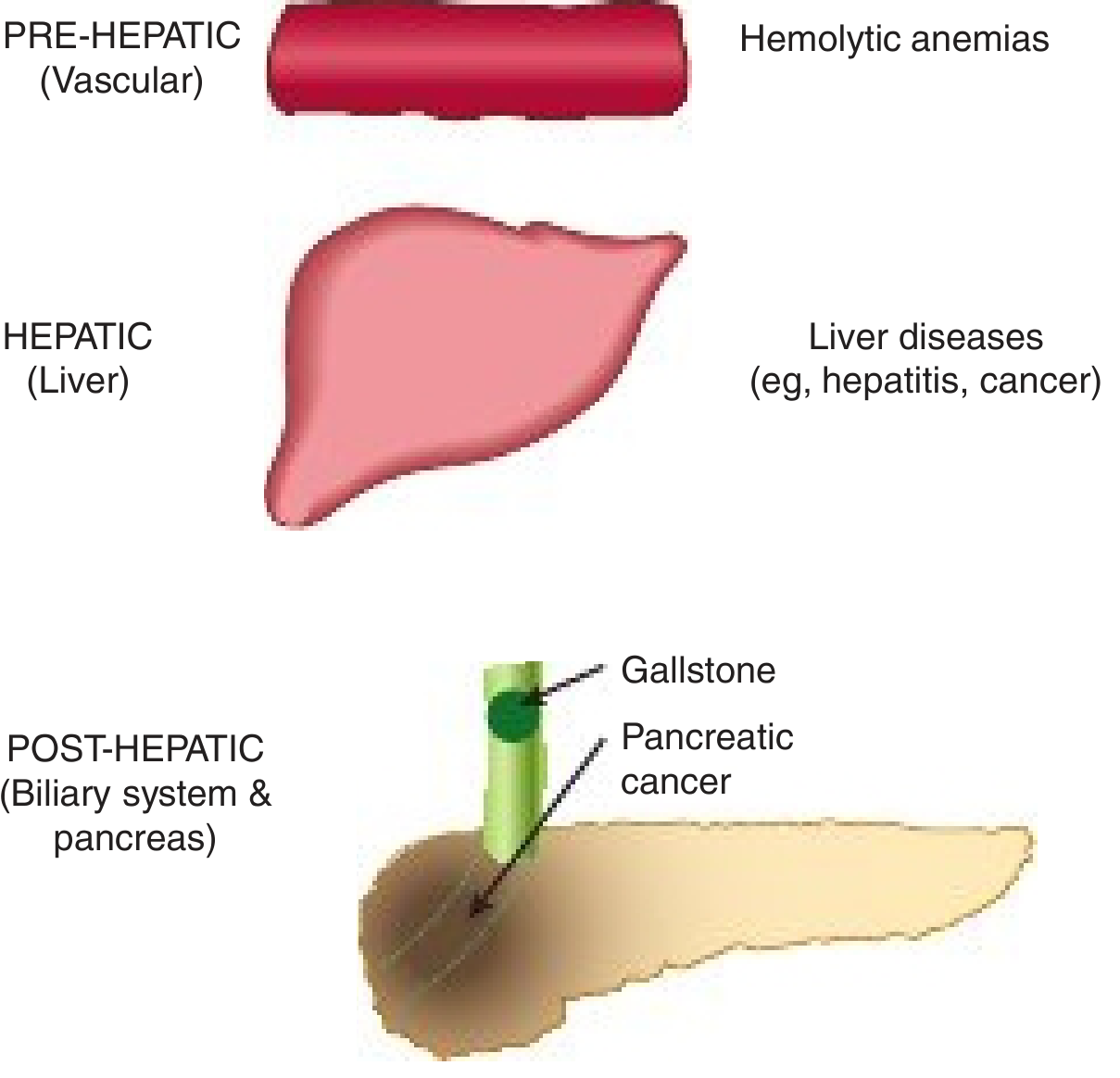
1. Pre-hepatic (Haemolytic) Jaundice
- Mechanism: Excessive red cell destruction → overproduction of unconjugated (indirect) bilirubin that overwhelms hepatic conjugation capacity
- Bilirubin type: ↑ Unconjugated (indirect)
- Causes: Hereditary spherocytosis, sickle cell disease, thalassemia, G6PD deficiency, immune haemolysis, malaria, massive blood transfusion, resorption of haematoma
- Serum bilirubin rarely exceeds 5 mg/dL in isolated haemolysis
- Urine: Urobilinogen ↑, no bilirubin (unconjugated is not water-soluble = acholuric jaundice)
- Stool: Urobilinogen ↑, dark stools
2. Hepatic (Hepatocellular) Jaundice
- Mechanism: Liver cell damage impairs both conjugation and excretion → mixed (conjugated + unconjugated) hyperbilirubinemia
- Bilirubin type: ↑ Both direct and indirect
- Causes: Viral hepatitis (A, B, C, E), alcoholic/non-alcoholic liver disease, drug toxicity (paracetamol, isoniazid), cirrhosis, autoimmune hepatitis, Wilson disease
- Urine: Bilirubin present (if micro-obstruction occurs), urobilinogen variable
- Stool: Pale (reduced urobilinogen)
- ALT/AST elevated out of proportion to alkaline phosphatase
3. Post-hepatic (Obstructive / Cholestatic) Jaundice
- Mechanism: Blockage of the biliary tree prevents conjugated bilirubin from reaching the intestine → it regurgitates into blood
- Bilirubin type: ↑ Conjugated (direct)
- Causes: Gallstones in common bile duct, carcinoma of the head of the pancreas, cholangiocarcinoma, primary sclerosing cholangitis
- Urine: Bilirubin present (dark urine = choluric jaundice), no urobilinogen (bile cannot reach gut)
- Stool: Pale/clay-coloured (absent urobilinogen)
- Alkaline phosphatase elevated out of proportion to ALT/AST
Lab Comparison Table
| Normal | Pre-hepatic | Hepatic | Post-hepatic | |
|---|---|---|---|---|
| Serum bilirubin | Direct 0.1–0.4, Indirect 0.2–0.7 mg/dL | ↑ Indirect | ↑ Both | ↑ Direct |
| Urine bilirubin | Absent | Absent | Present (if obstruction) | Present |
| Urine urobilinogen | 0–4 mg/24h | Increased | Decreased | Absent |
| Fecal urobilinogen | 40–280 mg/24h | Increased | Decreased | Trace/absent |
— Harper's Illustrated Biochemistry, 32nd ed.
Diagnostic Approach
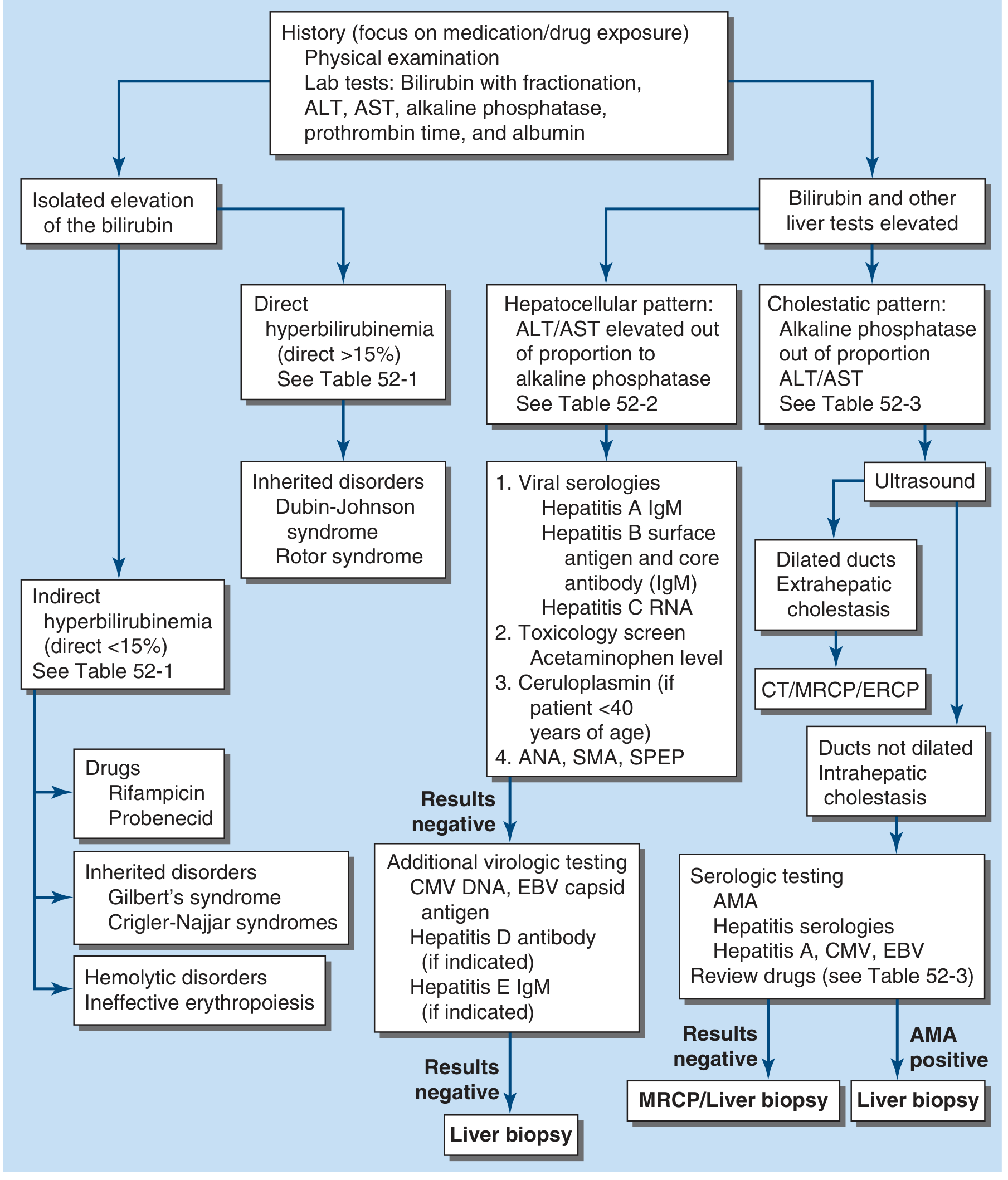
Key first step: fractionate bilirubin (direct vs. indirect) and check ALT, AST, alkaline phosphatase, and albumin. — Harrison's, 22nd ed.
Gilbert Syndrome
Definition and Genetics
Gilbert syndrome is the most common hereditary hyperbilirubinemia, affecting 3–7% of the population (male:female ratio ~1.5–7:1). It results from a promoter polymorphism in the UGT1A1 gene (UDP-glucuronosyltransferase 1A1).
- The most common variant is the UGT1A1*28 allele — a homozygous A(TA)₂TAA → A(TA)₇TAA insertion in the TATA box of the UGT1A1 promoter, which reduces transcription factor binding and promoter activity
- Allele frequency: ~42% in Black, ~30% in White, ~10% in Asian populations (in Asians, coding region mutations are more common than the *28 allele)
- Results in ~10–35% of normal UGT1A1 (bilirubin UDPGT) activity — enough to maintain near-normal bilirubin most of the time
- Approximately 4–16% of the population are homozygous carriers
— Goldman-Cecil Medicine; Yamada's Textbook of Gastroenterology, 7th ed.; Harrison's, 22nd ed.
Pathophysiology
Reduced UGT1A1 activity impairs conjugation of unconjugated bilirubin → mild unconjugated (indirect) hyperbilirubinemia. The condition typically first presents in adolescence, when increased sex steroids alter bilirubin metabolism.
Clinical Features
- Benign and asymptomatic — no liver disease, no structural hepatic abnormality
- Serum bilirubin usually < 3 mg/dL (< 6 mg/dL at most) — unconjugated
- Jaundice is intermittent, triggered by:
- Fasting / caloric restriction
- Intercurrent illness / fever
- Physical or emotional stress
- Alcohol use
- Surgery
- Phenotypic distinction between mild Gilbert syndrome and a normal bilirubin level is often blurred
Comparison with Related Conjugation Disorders
| Condition | Enzyme Activity | Bilirubin Level | Prognosis |
|---|---|---|---|
| Gilbert syndrome | ~10–35% of normal | < 6 mg/dL (usually < 3) | Benign; normal life expectancy |
| Crigler-Najjar Type II | < 10% | 6–25 mg/dL | Usually survive to adulthood; kernicterus risk under stress |
| Crigler-Najjar Type I | 0% (absent) | > 20 mg/dL | Fatal in infancy without treatment (phototherapy/liver transplant) |
— Harrison's 22nd ed.; Quick Compendium of Clinical Pathology, 5th ed.
Important Drug Interaction — Irinotecan
A critical clinical exception: the antitumour drug irinotecan (CPT-11) has its active metabolite SN-38 glucuronidated specifically by UGT1A1. In Gilbert syndrome patients, reduced UGT1A1 activity leads to SN-38 accumulation → severe toxicity including:
- Intractable diarrhoea
- Myelosuppression
- Other serious adverse effects
Raloxifene (oestrogen modulator) is also a UGT1A1 substrate; homozygous *28 carriers have ~2-fold higher drug exposure.
Phenobarbital induces hepatic enzyme activity and normalises bilirubin in Gilbert syndrome (as well as Crigler-Najjar type II). — Goldman-Cecil Medicine
Conjugated Hyperbilirubinemia: Related Hereditary Syndromes
| Syndrome | Defect | Bilirubin Type | Feature |
|---|---|---|---|
| Dubin-Johnson | MRP2 (ABCC2) mutation → impaired canalicular bilirubin export | Conjugated | Heavily pigmented liver; otherwise normal LFTs |
| Rotor syndrome | OATP1B1 + OATP1B3 mutation → impaired sinusoidal reuptake | Conjugated | No hepatic pigmentation; ↑ total urinary coproporphyrin |
Both are benign with good prognosis. — Harrison's 22nd ed.; Yamada's, 7th ed.
Key takeaway: Gilbert syndrome sits at the mildest end of the UGT1A1 deficiency spectrum — it is a clinical curiosity rather than a disease, but is important to recognise to avoid unnecessary investigation and to flag the irinotecan drug interaction.
This is a shared conversation. Sign in to Orris to start your own chat.