I now have comprehensive material from multiple authoritative sources. Let me compose the full structured answer for all 14 questions.
Anemia — Complete Study Guide (Questions 53–66)
53. Modern Classification of Anemia
There are two complementary classification systems:
A. By Red Cell Size (MCV-Based) — Wintrobe Classification
| Type | MCV | Causes |
|---|
| Microcytic | < 80 fL | Iron deficiency, thalassemia, anemia of inflammation, sideroblastic anemia |
| Normocytic | 80–100 fL | Aplastic anemia, renal disease, endocrinopathies, myeloma, early iron/B12 deficiency |
| Macrocytic | > 100 fL | B12/folate deficiency (oval macrocytes); alcohol, liver disease, hypothyroidism (round macrocytes) |
B. By Mechanism (Reticulocyte-Based)
Hyperproductive (high reticulocyte count):
- Bleeding (hemorrhage)
- Hemolysis — acquired (autoimmune, mechanical) or congenital (enzyme defects, hemoglobinopathies, membrane defects)
Hypoproductive (low/normal reticulocyte count):
- Nutritional deficiency: iron, B12, folate, copper, vitamin C
- Absence of red cell precursors: aplastic anemia, pure red cell aplasia
- Lack of erythropoietin: renal disease, anemia of inflammation, aging
- Marrow replacement: neoplasm, infection, granulomatous disease
- Stem cell defects: myelodysplasia, leukemia
Anemia is defined as blood counts below normal for a given population. — Harrison's Principles of Internal Medicine 22E, p. 492
54. Etiological Factors Contributing to Anemic Syndrome
Anemia arises from three fundamental mechanisms, each with multiple causes:
1. Decreased Red Cell Production
- Nutritional deficiency: Iron (most common worldwide), vitamin B12, folate
- Bone marrow suppression: Toxins, radiation, drugs (chemotherapy, chloramphenicol)
- Marrow replacement: Leukemia, lymphoma, metastatic carcinoma, myelofibrosis
- Reduced erythropoietin: Chronic kidney disease, endocrine disorders (hypothyroidism, Addison's disease)
- Chronic inflammation: Cytokine-mediated inhibition of iron reutilization and erythropoietin response
2. Increased Red Cell Destruction (Hemolysis)
- Intrinsic (congenital) RBC defects: membrane, enzyme, or hemoglobin abnormalities
- Extrinsic (acquired): autoimmune antibodies, infections, toxins, mechanical trauma, hypersplenism
3. Blood Loss
- Acute: Trauma, surgery, GI hemorrhage
- Chronic: GI bleeding (peptic ulcer, cancer, hookworm), menorrhagia, frequent phlebotomy
At least 75% of all cases of anemia are hypoproliferative in nature; the most common cause is mild to moderate iron deficiency or inflammation. — Harrison's 22E, p. 492
55. Classification of Hemolytic Anemias: Congenital and Acquired Forms
Classification by Site
- Intravascular hemolysis: Free hemoglobin released into bloodstream → hemoglobinemia, hemoglobinuria, acute renal failure risk (e.g., ABO incompatibility, G6PD in oxidative crisis)
- Extravascular hemolysis: RBCs removed by macrophages in spleen/liver → jaundice, splenomegaly (e.g., hereditary spherocytosis, warm AIHA)
Classification by Origin
INTRINSIC (Congenital) — Defects Inside the RBC
| Category | Examples |
|---|
| Membrane defects | Hereditary spherocytosis, hereditary elliptocytosis, PNH (acquired) |
| Enzyme defects | G6PD deficiency (X-linked; triggered by oxidant drugs/fava beans), Pyruvate kinase deficiency |
| Hemoglobin defects | Sickle cell anemia, thalassemias (α and β), HbC disease |
EXTRINSIC (Acquired) — Defects Outside the RBC
| Category | Examples |
|---|
| Immune-mediated | Warm AIHA (IgG, 37°C — idiopathic, SLE, lymphoma, drugs), Cold agglutinin disease (IgM — infections, lymphoma), Drug-induced |
| Microangiopathic | TTP, HUS, DIC, malignant hypertension, prosthetic heart valves |
| Infections | Malaria, Clostridium, Bartonella |
| Hypersplenism | Sequestration and destruction in enlarged spleen |
There are multiple classification systems for hemolytic anemias based on Coombs test reactivity, intrinsic vs. extrinsic defects, intravascular vs. extravascular hemolysis, and congenital or acquired forms. — Rosen's Emergency Medicine, p. 2446
56. Diagnostic Criteria for Iron Deficiency Anemia (IDA)
Clinical Features
- Fatigue, pallor, dyspnea on exertion (from anemia)
- Koilonychia (spoon nails), alopecia, atrophic glossitis, pica (craving for clay, ice)
- Esophageal webs (Plummer-Vinson syndrome in severe, chronic cases)
Laboratory Findings
| Test | Result in IDA |
|---|
| Hb/Hct | Decreased |
| MCV | Decreased (microcytic) |
| MCH/MCHC | Decreased (hypochromic) |
| Serum iron | Low |
| TIBC (Transferrin) | High (elevated) |
| Transferrin saturation | < 15% |
| Serum ferritin | Low (< 30 µg/L = iron store depletion) |
| Reticulocytes | Low/normal (hypoproliferative) |
| Hepcidin | Low |
Peripheral Blood Smear
- Microcytes, hypochromic cells with enlarged central pallor
- "Pencil cells" (elongated poikilocytes)
Bone Marrow
- Absent stainable iron (Prussian blue stain — most definitive finding)
- Mild to moderate erythroid hyperplasia
Response to Treatment
- Reticulocytosis within 5–7 days of oral iron
- Steady rise in Hb over 4–8 weeks
The diagnosis of iron deficiency anemia ultimately rests on laboratory studies: low serum iron, low ferritin, elevated TIBC, transferrin saturation below 15%. — Robbins & Kumar Pathology, p. 615
Iron deficiency anemia: hypochromic microcytic red cells with narrow rim of peripheral hemoglobin. — Robbins & Kumar Pathology
57. Diagnostic Criteria for B12 and Folate Deficiency Anemia
Both cause megaloblastic anemia — impaired DNA synthesis → delayed nuclear maturation with normal cytoplasmic maturation.
Clinical Features
| Feature | B12 Deficiency | Folate Deficiency |
|---|
| Anemia symptoms | Yes | Yes |
| Neurological | Subacute combined degeneration (dorsal + lateral columns): paresthesias, ataxia, loss of proprioception, spasticity | Neuropsychiatric (depression, forgetfulness) — no spinal cord involvement |
| Glossitis | Yes | Yes |
Laboratory Findings
| Test | Result |
|---|
| MCV | Elevated (macrocytic > 100 fL) |
| Peripheral smear | Macro-ovalocytes, hypersegmented neutrophils (≥5 lobes), anisocytosis, poikilocytosis |
| Reticulocyte count | Low |
| Serum B12 | Low (< 200 pg/mL) in B12 deficiency |
| Serum/RBC folate | Low in folate deficiency |
| Homocysteine | Elevated in both |
| Methylmalonic acid | Elevated in B12 deficiency; normal in folate deficiency ← key differentiator |
| Bone marrow | Hypercellular; megaloblasts at all erythroid stages; giant metamyelocytes |
| LDH | Elevated (ineffective erythropoiesis) |
| Bilirubin | Mildly elevated (intramedullary hemolysis) |
Differentiating B12 from Folate Deficiency
- Elevated methylmalonic acid → B12 deficiency (not folate)
- Normal methylmalonic acid + low folate → folate deficiency
- Neurologic findings → B12 deficiency only
- Body stores: B12 stores last 3–5 years; folate stores last only 2–4 months
The hematologic findings are identical in B12 and folate deficiency. Methylmalonic acid is elevated in B12 but not folate deficiency. Neurologic changes do not occur with folate deficiency alone. — Robbins & Kumar, p. 612
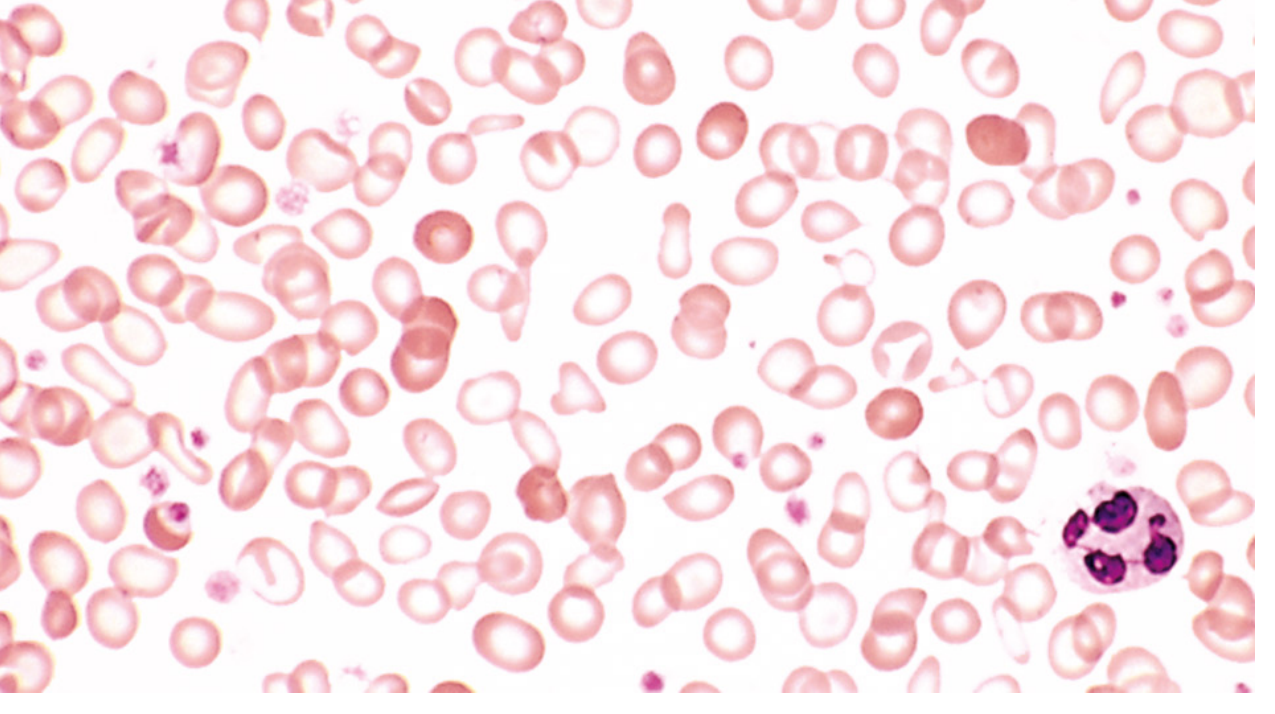
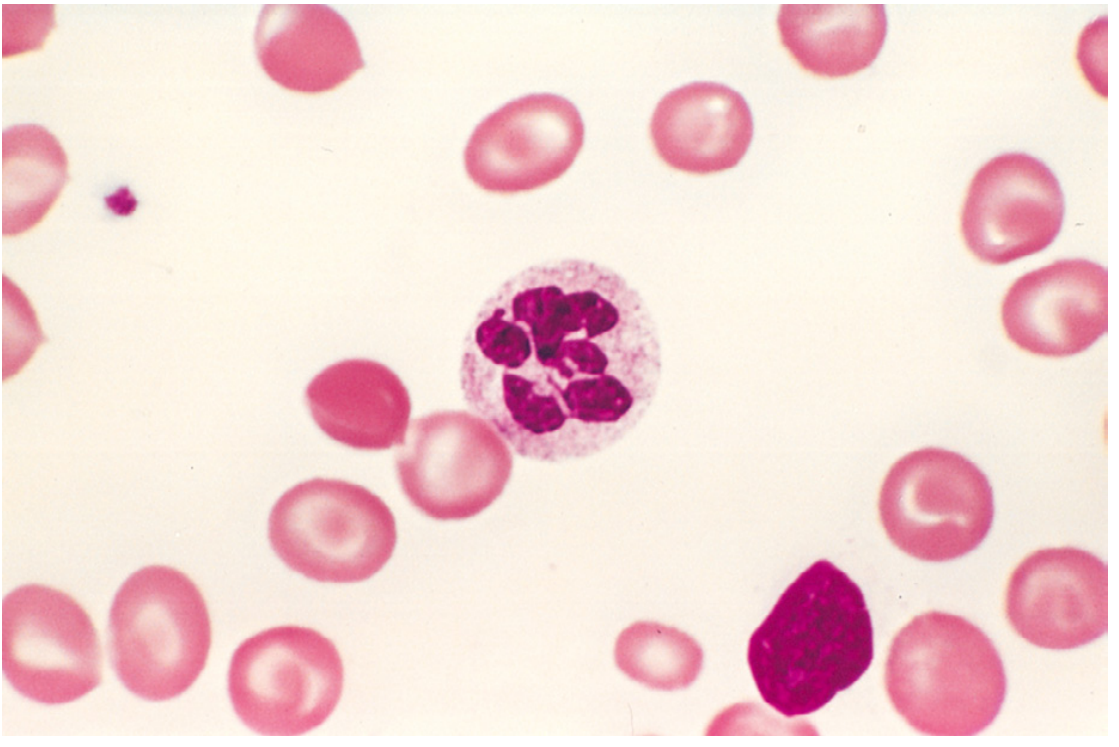
Megaloblastic anemia: peripheral blood smear with hypersegmented neutrophil (6-lobed nucleus). — Robbins & Kumar Pathology
58. Diagnostic Criteria for Hypo- and Aplastic Anemia
Aplastic anemia is characterized by pancytopenia due to destruction or suppression of hematopoietic stem cells, resulting in a hypocellular marrow replaced by fat.
Clinical Features
- Symptoms of anemia: fatigue, pallor, dyspnea
- Thrombocytopenia: petechiae, purpura, mucosal bleeding
- Neutropenia: recurrent infections
Diagnostic Criteria (Severe Aplastic Anemia — International AA Study Group)
Peripheral blood — at least 2 of 3:
- Neutrophils < 0.5 × 10⁹/L
- Platelets < 20 × 10⁹/L
- Reticulocytes < 20 × 10⁹/L (< 1%)
PLUS bone marrow biopsy:
- Cellularity < 25% (or < 50% with < 30% residual hematopoietic cells)
- Replacement of marrow space by fat cells
Laboratory Features
- Pancytopenia (all cell lines reduced)
- Normocytic or mildly macrocytic anemia
- Low/absent reticulocytes
- Normal RBC morphology (no dysplasia)
- Normal or elevated MCV
- Elevated erythropoietin
Bone Marrow (critical)
- Hypocellular or acellular marrow biopsy
- Fat cell replacement
- No dysplasia, no blasts, no fibrosis
- Prussian blue: absent iron in aplasia vs present in MDS
59. Diagnostic Criteria for Hemolytic Anemia
General Diagnostic Criteria
Evidence of accelerated RBC destruction:
| Test | Finding |
|---|
| Serum LDH | Elevated (released from lysed RBCs) |
| Serum haptoglobin | Low (binds free Hb, depleted) |
| Indirect bilirubin | Elevated (Hb catabolism) |
| Urobilinogen | Elevated in urine/stool |
| Free plasma hemoglobin | Elevated (intravascular) |
| Hemoglobinuria | Present in intravascular hemolysis |
| Hemosiderinuria | Chronic intravascular hemolysis |
Evidence of compensatory increased production:
- Reticulocytosis (elevated reticulocyte count)
- Bone marrow erythroid hyperplasia (↑ M:E ratio inversion)
Peripheral Blood Smear Findings (type-specific):
- Spherocytes → hereditary spherocytosis, warm AIHA
- Schistocytes (helmet cells) → MAHA (TTP, DIC, prosthetic valves)
- Sickle cells → sickle cell disease
- Target cells → HbC, thalassemia, liver disease
Specific Tests:
- Direct Coombs (DAT): Positive in immune-mediated HA → confirms autoantibody/complement on RBC surface
- Indirect Coombs: Characterizes antibody type/target
- Osmotic fragility test: Hereditary spherocytosis
- Hb electrophoresis: Hemoglobinopathies
- G6PD enzyme assay: G6PD deficiency
- Flow cytometry (CD55/CD59 deficiency): PNH
60. Differential Diagnosis: Hypo-Aplastic Anemia vs. Leukemia
Both may present with pancytopenia, but they differ fundamentally:
| Feature | Aplastic Anemia | Acute Leukemia |
|---|
| Onset | Gradual or subacute | Often acute/subacute |
| Organomegaly | Absent | Hepatosplenomegaly, lymphadenopathy common |
| Lymph nodes | Normal | Often enlarged |
| WBC | Low (leukopenia) | Normal, low, or markedly elevated |
| Peripheral smear | Pancytopenia, normal cell morphology, no blasts | Blasts present (> 20% = AML) |
| Bone marrow cellularity | Hypocellular (< 25%), replaced by fat | Hypercellular, packed with blast cells |
| Blasts in marrow | < 5% (absent/rare) | ≥ 20% (AML) or lymphoblasts (ALL) |
| Chromosome analysis | Normal karyotype (usually) | Chromosomal abnormalities common |
| LDH | Normal to mildly elevated | Often markedly elevated |
| Uric acid | Normal | Often elevated (high cell turnover) |
| Response to IST | Responds to immunosuppression/ATG | Does not respond; requires chemotherapy |
| Trephine biopsy | Fat spaces, no malignant infiltrate | Effacement of marrow architecture by blasts |
Key distinguishing feature: bone marrow biopsy. Aplastic anemia shows a hypocellular marrow with fat replacement and no blasts; leukemia shows a hypercellular marrow packed with malignant blast cells. — Rosen's Emergency Medicine
61. Laboratory Indicators for Hypo- and Aplastic Anemia
CBC and Peripheral Blood
- Anemia: Normocytic (MCV normal or slightly elevated), normochromic
- Leukopenia: Particularly neutropenia (< 1.5 × 10⁹/L in mild; < 0.5 × 10⁹/L in severe)
- Thrombocytopenia: Platelet count < 50–20 × 10⁹/L
- Reticulocyte count: Very low (< 20 × 10⁹/L) — critical finding
- No blasts, no dysplastic changes on smear
Other Tests
- Erythropoietin: Elevated (compensatory)
- Iron studies: Normal or elevated serum iron; normal to elevated ferritin (no utilization)
- Serum B12/folate: Normal
- Liver function tests: Hepatitis serology (post-hepatitis aplastic anemia)
- Flow cytometry (PNH clone): CD55/CD59 negative population
- Chromosomal analysis (peripheral blood): For Fanconi anemia (chromosomal breakage)
Bone Marrow Aspirate and Biopsy
- Aspirate: "Dry tap" or very hypocellular sample; fatty stroma
- Biopsy (trephine): Definitive — hypocellular marrow (< 25–30% cellularity), fat replacement, residual lymphocytes and plasma cells, absence of hematopoietic precursors
- No dysplasia, no fibrosis, no blasts
62. Methods of Bone Marrow Examination
1. Bone Marrow Aspiration (BMA)
- Site: Posterior iliac crest (preferred), sternum (sternal aspiration — emergency use), anterior iliac crest, tibial bone (in infants)
- Procedure: Needle aspiration of marrow fluid; smears prepared and stained (Giemsa/Wright, Perls' Prussian blue for iron, PAS, Sudan black)
- Yields: Cytology of individual cells — morphology of erythroid, myeloid, megakaryocytic series; M:E ratio; blast percentage; iron stores
- Limitations: May be a "dry tap" in fibrosis or aplasia; does not assess overall architecture
2. Bone Marrow Biopsy (Trephine Biopsy)
- Site: Posterior superior iliac spine
- Procedure: Core biopsy taken with a Jamshidi or Westerman-Jensen needle; formalin-fixed, paraffin-embedded; sections stained with H&E, reticulin, iron
- Yields: Marrow cellularity, architecture, presence/extent of fibrosis, infiltration (tumor, granulomas), megakaryocyte distribution
- Essential for: Aplastic anemia, myelofibrosis, lymphoma staging, hypocellular states, "dry tap"
3. Special Stains on Marrow Preparations
| Stain | Purpose |
|---|
| Prussian Blue (Perls') | Iron stores (sideroblasts, hemosiderin) |
| PAS (periodic acid-Schiff) | Glycogen (lymphoblasts in ALL) |
| Reticulin/Masson's trichrome | Fibrosis grading |
| Sudan Black B / MPO | Myeloid differentiation (AML) |
4. Ancillary Studies
- Flow cytometry: Immunophenotyping (leukemias, PNH clones)
- Cytogenetics/FISH: Chromosomal abnormalities (leukemia, MDS)
- Molecular studies: JAK2, BCR-ABL, FLT3, NPM1, etc.
- Culture: For infection (tuberculosis, fungal)
63. Diagnosis and Principles of Treatment of Iron Deficiency Anemia
Diagnosis (Summary)
- History: chronic blood loss (GI, menorrhagia), dietary insufficiency, malabsorption, pregnancy
- CBC: microcytic hypochromic anemia
- Iron studies: ↓ serum iron, ↓ ferritin, ↑ TIBC, transferrin saturation < 15%
- Peripheral smear: microcytes, hypochromia, pencil cells
- In adult males/postmenopausal females: must exclude GI malignancy as source
Principles of Treatment
1. Identify and treat the underlying cause (most important step)
- Treat GI bleeding source, fibroids, malabsorption, dietary deficiency
2. Iron supplementation
Oral iron (first-line):
- Ferrous sulfate 325 mg TID (65 mg elemental iron/dose) — standard
- Take on empty stomach for best absorption; vitamin C enhances absorption
- Expected response: reticulocytosis in 5–7 days; Hb rises ~1–2 g/dL per 2–3 weeks
- Treat for 3–6 months after Hb normalization to replenish stores
- Side effects: nausea, constipation, dark stools
Parenteral iron (IV/IM — indications):
- Malabsorption (celiac disease, gastric bypass)
- Intolerance to oral iron
- Need for rapid repletion (severe anemia, chronic dialysis)
- Agents: iron sucrose, ferric gluconate, ferric carboxymaltose, low-molecular-weight iron dextran
3. Blood transfusion
- Reserved for severe symptomatic anemia (Hb < 7 g/dL) or hemodynamic instability
4. Dietary counseling
- Increase dietary heme iron (red meat, poultry, fish)
- Avoid tea/coffee with meals (inhibit absorption)
64. Iron-Containing Drugs: Indications and Contraindications
Oral Iron Preparations
| Drug | Elemental Iron Content |
|---|
| Ferrous sulfate | 20% (65 mg per 325 mg tablet) |
| Ferrous gluconate | 12% (36 mg per 300 mg tablet) |
| Ferrous fumarate | 33% (99 mg per 300 mg tablet) |
| Ferric sulfate (Vitron-C) | Various |
| Polysaccharide-iron complex | Variable |
Indications: Iron deficiency anemia, iron deficiency without anemia (prevention), pregnancy supplementation, infants on cow's milk diet
Contraindications (oral):
- Hemochromatosis / hemosiderosis (iron overload)
- Hemolytic anemia without iron deficiency (can worsen iron overload)
- Active inflammatory bowel disease (may worsen GI irritation)
- Repeated transfusions (risk of secondary iron overload)
- Thalassemia (unless IDA confirmed)
Drug Interactions:
- Reduces absorption of: fluoroquinolones, tetracyclines, levothyroxine, levodopa, bisphosphonates (separate by 2–4 hours)
- Absorption decreased by: antacids, calcium, coffee, tea, PPI therapy
Parenteral Iron Preparations
| Drug | Route | Notes |
|---|
| Iron sucrose (Venofer) | IV | Safe, widely used; in dialysis patients |
| Ferric gluconate (Ferrlecit) | IV | Used in CKD/hemodialysis |
| Ferric carboxymaltose (Injectafer) | IV | High-dose single infusion possible |
| Iron dextran (low-MW) | IV/IM | Test dose required; anaphylaxis risk |
| Ferumoxytol | IV | Rapid infusion; MRI interference |
Indications for parenteral iron:
- Oral intolerance/non-compliance
- Malabsorption syndromes
- Active/ongoing blood loss exceeding oral replenishment
- Chronic kidney disease (on dialysis or with EPO therapy)
- Inflammatory bowel disease
- Perioperative anemia optimization
- Heart failure with iron deficiency (even without anemia)
Contraindications (parenteral):
- Iron overload states
- Acute infection (iron may promote bacterial growth)
- Known hypersensitivity to preparation
- First trimester of pregnancy (for most preparations)
65. Diagnosis and Principles of Treatment of B12 and Folate Deficiency Anemia
Diagnosis
- Macrocytic anemia (MCV > 100 fL)
- Peripheral smear: macro-ovalocytes, hypersegmented neutrophils (≥ 5 lobes)
- Low serum B12 (< 200 pg/mL) or low serum/RBC folate
- Elevated homocysteine (both); elevated methylmalonic acid (B12 only)
- Bone marrow: megaloblastic changes, hypercellularity
- Pernicious anemia (main cause of B12 deficiency): anti-intrinsic factor antibodies (specific), anti-parietal cell antibodies (sensitive); Schilling test (historical)
- Rule out B12 deficiency before starting folate — folate therapy corrects hematologic findings but does NOT prevent and may worsen neurologic deterioration from B12 deficiency
Treatment: B12 Deficiency
- Cause of B12 deficiency determines route:
- Dietary deficiency: oral B12 1,000 µg/day (high-dose oral can compensate for lack of intrinsic factor via passive diffusion)
- Pernicious anemia / malabsorption: IM/SC cyanocobalamin or hydroxocobalamin 1,000 µg daily × 7 days → weekly × 4 weeks → monthly (lifelong)
- Neurologic improvement may occur up to 6–12 months with treatment; subacute combined degeneration may be irreversible if longstanding
- Reticulocytosis peaks at 5–7 days; Hb normalizes over 1–2 months
Treatment: Folate Deficiency
- Oral folic acid 1–5 mg/day for 4 months (until stores replenished)
- Treat underlying cause: alcohol cessation, manage malabsorption
- Pregnancy prophylaxis: 0.4–0.8 mg/day (reduces neural tube defects)
- Folinic acid (leucovorin) for drug-induced folate deficiency (methotrexate, trimethoprim)
66. Diagnosis and Principles of Treatment of Hemolytic Anemia
Diagnosis
Step 1 — Confirm hemolysis:
- ↑ LDH, ↓ haptoglobin, ↑ indirect bilirubin, ↑ reticulocytes
- Hemoglobinuria/hemosiderinuria (intravascular)
Step 2 — Characterize the hemolysis:
- Direct Coombs test (DAT): If positive → immune-mediated (AIHA, drug-induced)
- Peripheral smear: Spherocytes (AIHA, hereditary spherocytosis), schistocytes (MAHA), sickle cells, target cells
- Osmotic fragility: Hereditary spherocytosis
- Hb electrophoresis: Sickle cell, thalassemia, HbC
- G6PD assay: G6PD deficiency (sample after acute episode when reticulocytes normalized)
- Flow cytometry (CD55/CD59): PNH
- Bone marrow: Erythroid hyperplasia; for PNH/aplastic context
Principles of Treatment
Autoimmune Hemolytic Anemia (Warm AIHA):
- Corticosteroids (first-line): Prednisolone 1 mg/kg/day → taper over months
- Splenectomy: Second-line if steroid failure/dependence
- Rituximab (anti-CD20): Effective for refractory cases
- Immunosuppressants: Azathioprine, cyclophosphamide (third-line)
- Blood transfusion: Only for life-threatening anemia (cross-matching is difficult)
- Treat underlying cause: Lymphoma, SLE, drugs
Cold Agglutinin Disease:
- Avoidance of cold
- Rituximab (most effective)
- Plasmapheresis (acute severe)
- Steroids less effective
G6PD Deficiency:
- Avoid oxidant triggers (drugs — see Box above, fava beans, infections)
- Supportive: hydration, transfusion in acute crises
- No enzyme replacement available
Hereditary Spherocytosis:
- Splenectomy (curative for hemolysis — removes main destruction site)
- Folic acid supplementation (chronic hemolysis increases folate demand)
- Cholecystectomy if gallstones
Sickle Cell Disease:
- Hydroxyurea (increases HbF production)
- Prophylactic penicillin + pneumococcal vaccination
- Chronic transfusion (stroke prevention)
- Hematopoietic stem cell transplantation (potentially curative)
- Voxelotor, crizanlizumab (newer agents)
PNH:
- Eculizumab/ravulizumab (anti-C5 complement inhibitors) — reduces hemolysis and thrombosis
- Hematopoietic stem cell transplantation (only cure)
- Anticoagulation (thrombosis prevention)
- Meningococcal vaccination required before C5 inhibitor therapy
Aplastic Anemia with Hemolysis (PNH clone):
- Immunosuppression with anti-thymocyte globulin (ATG) + cyclosporine
- Eltrombopag (TPO receptor agonist)
- HSCT for severe cases in young patients
Sources: Harrison's Principles of Internal Medicine 22E | Robbins & Kumar Pathology (10e) | Rosen's Emergency Medicine