Description aetiology pathogenesis and clinical feature of hyaline a membrane disease and acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the content and images needed. Let me compose the comprehensive answer.
Hyaline Membrane Disease (Neonatal RDS) and Acute Respiratory Distress Syndrome (ARDS)
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.); Robbins & Kumar - Basic Pathology
PART 1: HYALINE MEMBRANE DISEASE (HMD) / Neonatal Respiratory Distress Syndrome (Neonatal RDS)
Description
Neonatal RDS, also called hyaline membrane disease (HMD), is the most common cause of respiratory distress in the newborn. The name derives from the deposition of a layer of hyaline proteinaceous material in the peripheral airspaces of affected infants. It is fundamentally a disease of prematurity - incidence increases sharply with decreasing gestational age:
| Gestational Age | Incidence |
|---|---|
| 37 weeks | ~1% |
| 34 weeks | ~10.5% |
| ≤28 weeks (without treatment) | ~93% |
Aetiology
The primary cause is pulmonary immaturity leading to surfactant deficiency. Key aetiological factors include:
- Prematurity (most common) - type II pneumocytes are insufficiently mature to produce adequate surfactant; surfactant production accelerates after the 35th week of gestation
- Male sex - strong association
- Maternal diabetes - elevated fetal insulin levels counteract the surfactant-stimulating effect of corticosteroids
- Caesarean delivery - bypasses the stress-induced cortisol surge that normally stimulates surfactant maturation
- Genetic mutations - congenital deficiency of SP-B (SFTPB gene) or SP-C (SFTPC gene) causes severe respiratory failure through absent surfactant proteins
- Second twin - higher risk than the first born twin
Conditions that reduce risk: intrauterine stress, fetal growth restriction (FGR) - these raise cortisol levels that accelerate lung maturation.
Pathogenesis
The core defect is insufficient pulmonary surfactant. Normal surfactant consists of:
- Dipalmitoyl phosphatidylcholine (lecithin) - the main lipid component
- Phosphatidylglycerol - smaller amounts
- Hydrophilic proteins SP-A and SP-D - pulmonary host defense
- Hydrophobic proteins SP-B and SP-C - reduce surface tension at the air-liquid interface
The cascade of events is illustrated below:
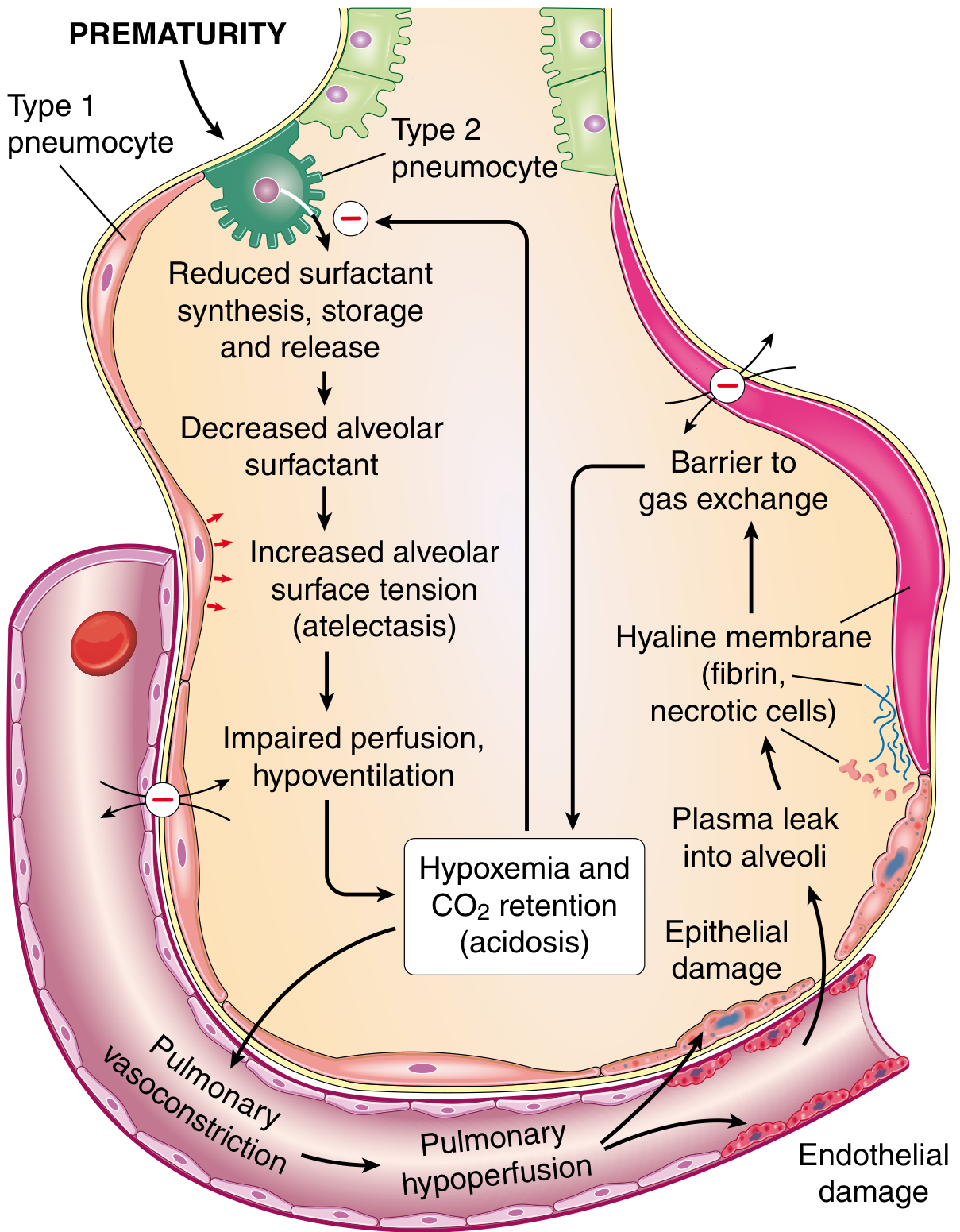
Step-by-step pathogenesis:
- Prematurity → reduced surfactant synthesis/storage/release by immature type II pneumocytes
- Decreased alveolar surfactant → increased alveolar surface tension
- Increased surface tension → alveolar collapse (atelectasis) with each expiration - the soft thoracic wall is pulled inward as the diaphragm descends, worsening the problem
- Atelectasis → impaired perfusion and hypoventilation → V/Q mismatch
- Hypoxemia and CO2 retention (acidosis)
- Hypoxemia further impairs surfactant synthesis - a vicious cycle
- Reduced lung compliance forces the infant to work as hard with every breath as with the first
- Endothelial and epithelial damage → plasma leaks into the alveoli
- Leaked protein-rich, fibrin-rich exudate + necrotic type II pneumocyte debris organize into eosinophilic hyaline membranes, lining the respiratory bronchioles and alveolar ducts
- Hyaline membranes act as a barrier to gas exchange, completing the vicious cycle
Hormonal modulation of surfactant synthesis: Cortisol, prolactin, thyroxine, TGF-β all promote surfactant production. Insulin suppresses it - explaining the higher risk in infants of diabetic mothers.
Morphology (Gross and Microscopic)
Gross:
- Lungs are normal in size but solid, airless, and reddish-purple (liver-like colour)
- Lungs typically sink in water (absent air)
Microscopy:
- Alveoli are poorly developed and collapsed
- Early: necrotic cellular debris in terminal bronchioles and alveolar ducts
- Necrotic material organizes into eosinophilic hyaline membranes lining respiratory bronchioles, alveolar ducts, and alveoli
- Membranes composed of fibrin + debris from necrotic type II pneumocytes
- Remarkable paucity of neutrophilic infiltration
- Cuboidal epithelial lining of remaining airspaces (sign of lung immaturity)
- Lesions are NEVER seen in stillborns (requires breathing attempts)
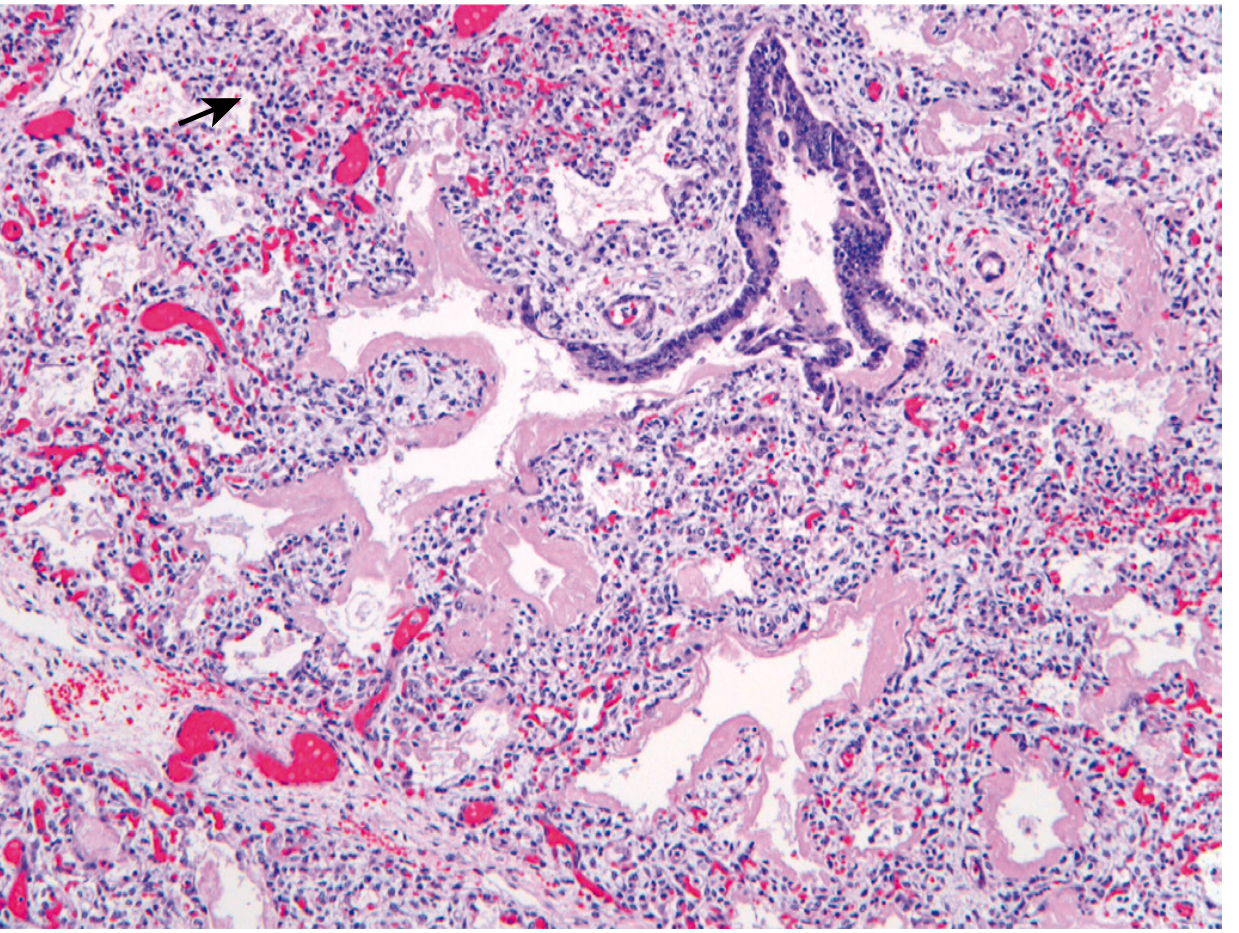
Reparative changes (after 48 hours survival):
- Alveolar epithelium proliferates under the membranes
- Membranes may detach and be phagocytosed by macrophages
- Alternatively, fibroblasts grow into membranes → incorporated into alveolar wall
Clinical Features
- Affected infants are almost always preterm with weight appropriate for gestational age
- Resuscitation may be needed at birth, but normal color/breathing often returns briefly
- Within 30 minutes, breathing becomes labored - grunting, nasal flaring, intercostal and subcostal retractions
- Within hours: cyanosis becomes evident in the untreated infant
- Fine rales over both lung fields
- Chest X-ray: uniform minute reticulogranular densities - classic "ground-glass" appearance with air bronchograms
- Full-blown condition: respiratory distress persists, cyanosis worsens; even 80% O2 via various ventilatory methods may fail
- If death is staved off for 3-4 days, the infant has an excellent chance of recovery (surfactant production matures)
Outcome and treatment:
- Prevention by antenatal corticosteroids (stimulate surfactant maturation)
- Amniotic fluid phospholipid analysis (lecithin:sphingomyelin ratio) assesses fetal lung maturity
- Exogenous surfactant therapy at birth for extremely premature infants (<28 weeks) has dramatically reduced mortality
- Complications include bronchopulmonary dysplasia (BPD), intraventricular hemorrhage, retinopathy of prematurity
PART 2: ACUTE RESPIRATORY DISTRESS SYNDROME (ARDS)
Description
ARDS (formerly also called Adult RDS) is a clinical syndrome of progressive respiratory insufficiency occurring in the setting of identifiable triggers such as sepsis, severe trauma, or diffuse pulmonary infection. It represents the severe end of a spectrum termed Acute Lung Injury (ALI).
ARDS is common in acutely ill patients: in a 2016 study across 50 countries, incidence was 10.4% in ICU patients, with mortality of 35% (mild), 40% (moderate), and 46% (severe). Approximately 190,000 cases occur per year in the United States.
Aetiology
ARDS can be triggered by direct (pulmonary) or indirect (extrapulmonary) lung injury:
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma with shock |
| Near-drowning | Burns |
| Inhalation of toxic gases | Acute pancreatitis |
| Lung contusion | Transfusion-related (TRALI) |
| Fat embolism | Drug overdose |
| Diffuse pulmonary infections | Cardiopulmonary bypass |
Risk factors: alcohol use disorder and cigarette smoking both worsen prognosis. Genetic variants in inflammation and coagulation pathways (identified by GWAS) increase susceptibility.
Pathogenesis
The central mechanism is diffuse alveolar damage (DAD) initiated by injury to both pneumocytes and pulmonary endothelium, setting in motion a self-amplifying inflammatory cascade:
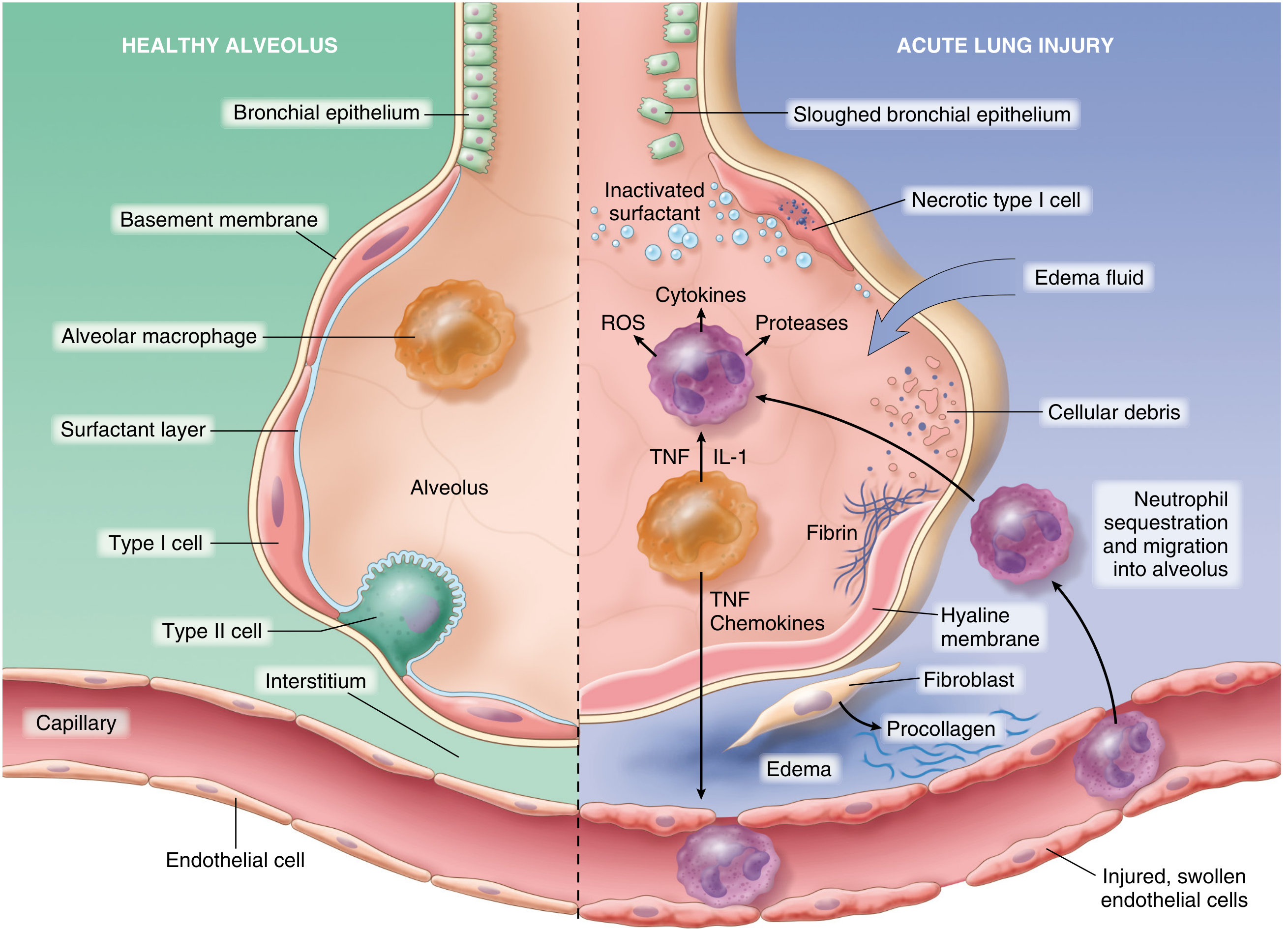
Step-by-step pathogenesis:
1. Initiating injury:
- Direct insult (e.g., inhaled toxin, aspiration) injures type I and type II pneumocytes
- Indirect insult (e.g., sepsis, circulating mediators) activates pulmonary endothelium
2. Endothelial activation (early event):
- Pneumocyte injury is sensed by resident alveolar macrophages, which secrete TNF acting on neighboring endothelium
- Circulating inflammatory mediators (sepsis/trauma) may activate endothelium directly
- Activated endothelium upregulates adhesion molecules, procoagulant proteins, and chemokines
3. Neutrophil recruitment:
- Neutrophils adhere to activated endothelium and migrate into interstitium and alveoli
- Degranulation releases proteases, reactive oxygen species (ROS), and cytokines
- Neutrophil extracellular traps (NETs) contribute directly to lung damage
- This creates a feedback loop of inflammation and endothelial damage
4. Vascular leak and hyaline membrane formation:
- Endothelial injury makes pulmonary capillaries leaky → interstitial and intraalveolar edema
- Type II pneumocyte necrosis → surfactant abnormalities → further impaired gas exchange
- Inspissated protein-rich edema fluid + dead alveolar epithelial debris → organize into hyaline membranes (the hallmark histologic lesion = diffuse alveolar damage)
5. V/Q mismatch:
- Lesions are not evenly distributed - stiff, poorly aerated regions coexist with near-normal areas
- Poorly aerated regions continue to be perfused → ventilation-perfusion mismatch → refractory hypoxemia
6. Resolution or fibrosis:
- If triggers abate: macrophages clear debris; fibrogenic cytokines (TGF-β, PDGF) stimulate fibroblasts → fibrosis of alveolar walls (fibro-proliferative stage)
- Type II pneumocytes proliferate to replace type I; endothelial restoration occurs
- In refractory cases: progressive cardiopulmonary failure
Morphology (Gross and Microscopic)
Gross (acute exudative stage):
- Lungs are heavy, firm, red, and boggy
- Congestion, interstitial and intraalveolar edema
Microscopy - Two phases:
| Phase | Histology |
|---|---|
| Acute exudative stage | Inflammation, fibrin deposition, diffuse alveolar damage; alveolar walls lined with waxy hyaline membranes (fibrin-rich edema + necrotic epithelial remnants) |
| Proliferative/organizing stage | Type II pneumocyte proliferation; granulation tissue in alveolar walls and spaces; increased immature fibroblasts in interstitium |
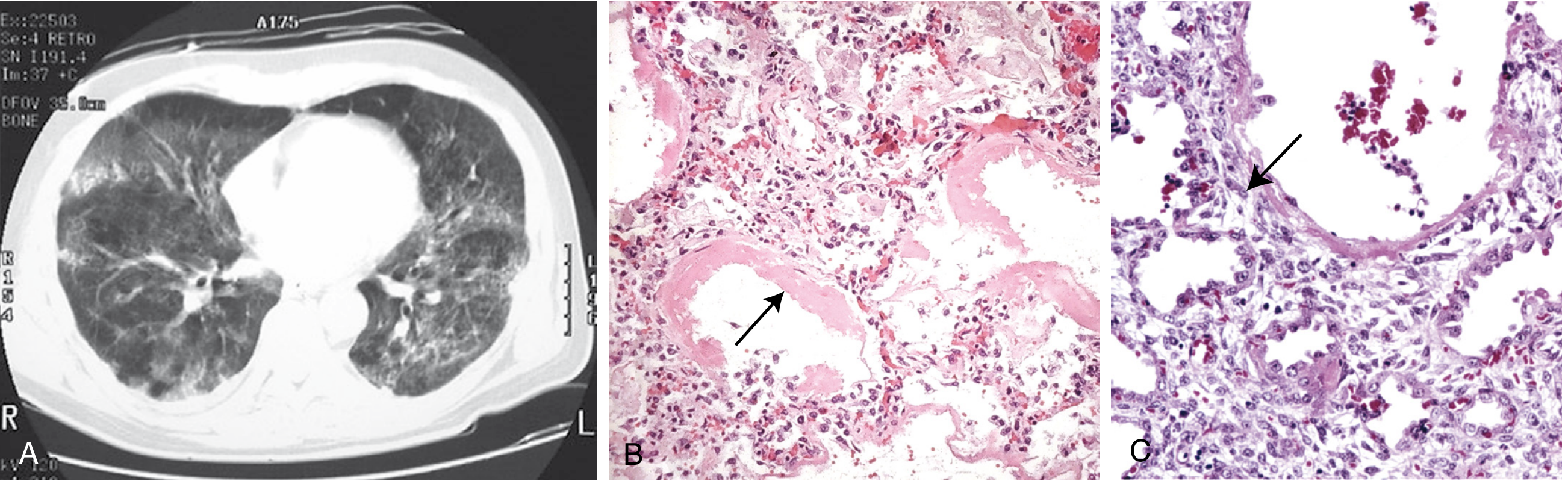
Clinical Features
Onset:
- Profound dyspnea and tachypnea are the heralding features, usually 12-48 hours after the precipitating event
- Rapidly progressive respiratory failure
Physical findings:
- Hypoxemia - often refractory to supplemental oxygen
- Cyanosis
- Diffuse bilateral crackles
- Tachycardia
Investigations:
- CXR/CT: bilateral diffuse opacities/infiltrates (ground-glass and consolidation) not explained by cardiac failure
- Arterial blood gas: hypoxemia (PaO2/FiO2 ratio <300 for ALI, <200 for ARDS); respiratory alkalosis early, respiratory acidosis late
- Berlin Definition (2012): bilateral opacities, respiratory failure within 1 week, not fully explained by cardiac failure/fluid overload, PaO2/FiO2 < 300 on PEEP ≥ 5 cmH2O
Course:
- Hypoxemia may be refractory due to V/Q mismatch
- Respiratory acidosis develops as disease progresses
- Lungs become stiff (loss of functional surfactant) → requires intubation, high ventilatory pressures
- No proven disease-specific treatment; management is supportive (lung-protective ventilation with low tidal volumes, prone positioning, PEEP)
- Most deaths attributable to sepsis, multiorgan failure, or severe lung injury
- Survivors usually recover pulmonary function; a minority develop interstitial fibrosis and chronic lung disease
KEY COMPARISON: HMD vs. ARDS
| Feature | HMD (Neonatal RDS) | ARDS |
|---|---|---|
| Population | Preterm neonates | Any age, usually adults |
| Primary defect | Surfactant deficiency (immaturity) | Diffuse alveolar damage from injury |
| Aetiology | Prematurity, maternal diabetes, C-section | Sepsis, trauma, aspiration, pneumonia |
| Pathogenesis | Surfactant deficiency → atelectasis → hypoxemia → hyaline membranes | Endothelial/epithelial injury → neutrophil recruitment → vascular leak → hyaline membranes |
| Gross | Liver-like, sinks in water | Heavy, firm, boggy, red |
| Histology | Collapsed alveoli + eosinophilic hyaline membranes; paucity of neutrophils | DAD: hyaline membranes + neutrophilic inflammation + fibrin; later type II hyperplasia + fibrosis |
| CXR | Fine reticulogranular "ground-glass" pattern | Bilateral diffuse opacities/consolidation |
| Key treatment | Exogenous surfactant; antenatal corticosteroids | Lung-protective ventilation; treat underlying cause |
| Prognosis | Excellent if survives 3-4 days | 35-46% mortality; survivors mostly recover function |
Both conditions share the hallmark of hyaline membrane formation, though via distinct mechanisms - surfactant insufficiency in HMD, and inflammatory alveolar damage in ARDS.
- Robbins, Cotran & Kumar - Pathologic Basis of Disease, 10th ed., Chapters 10 & 15
- Robbins & Kumar - Basic Pathology
Long cancer, how it is occur, classification, etiopathogenesis. Sıra sizde.
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the comprehensive material needed. Let me compose the full detailed answer.
Lung Cancer: Occurrence, Classification & Etiopathogenesis
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.); Harrison's Principles of Internal Medicine 22nd ed.
1. HOW LUNG CANCER OCCURS (Epidemiology & Overview)
Lung cancer is the leading cause of cancer-related death worldwide among both men and women. Key statistics:
- Over 2.2 million new cases globally in 2020; 1.8 million deaths per year
- In the United States: ~237,000 new cases and ~130,000 deaths annually (2022 estimates)
- Peak incidence in individuals in their fifties and sixties
- At diagnosis, >50% already have distant metastases; an additional ~25% have regional lymph node involvement
- Overall 5-year survival for all stages combined is only ~20%; even localized disease has only ~50% 5-year survival
- Since 2006, incidence has been declining ~2.5%/year in men and ~1%/year in women, attributed to reduced smoking, better screening, and improved therapies
The WHO defines lung cancer as tumors arising from the respiratory epithelium (bronchi, bronchioles, and alveoli).
2. CLASSIFICATION
2.1 Broad Clinical Division
Historically and clinically, lung cancers are divided into two major groups based on treatment approach:
| Category | Subtypes | % of Cases | Key Feature |
|---|---|---|---|
| Non-Small Cell Lung Cancer (NSCLC) | Adenocarcinoma, Squamous cell, Large cell | ~85% | More likely resectable; targeted therapy applicable |
| Small Cell Lung Cancer (SCLC) | Small cell carcinoma | ~15% | Almost always metastatic at presentation; chemo-sensitive but incurable |
2.2 WHO 2021 Classification (Histologic)
The 2021 WHO classification replaces the older SCLC/NSCLC binary. The simplified version is:
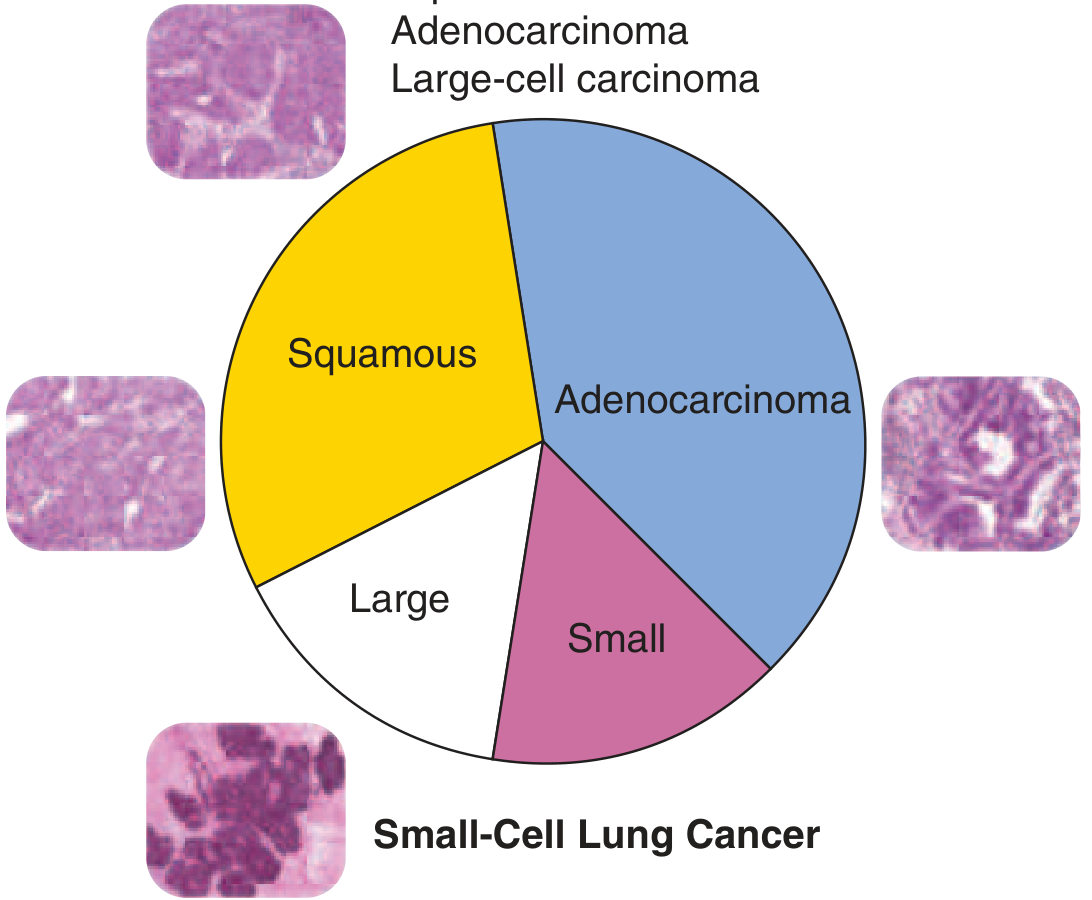
Table: WHO 2021 Classification of Malignant Epithelial Lung Tumors
| Type | Subtypes |
|---|---|
| Adenocarcinoma | Acinar, papillary, micropapillary, solid, lepidic predominant, mucinous |
| Squamous cell carcinoma | - |
| Large cell carcinoma | - |
| Neuroendocrine carcinomas | Small cell carcinoma, Large cell neuroendocrine carcinoma |
| Carcinoid tumors | Typical and atypical carcinoids |
| Mixed carcinomas | Adenosquamous, Small cell + other types |
| Other variants | Sarcomatoid, spindle cell, giant cell |
2.3 The Four Major Histologic Types (Detailed)
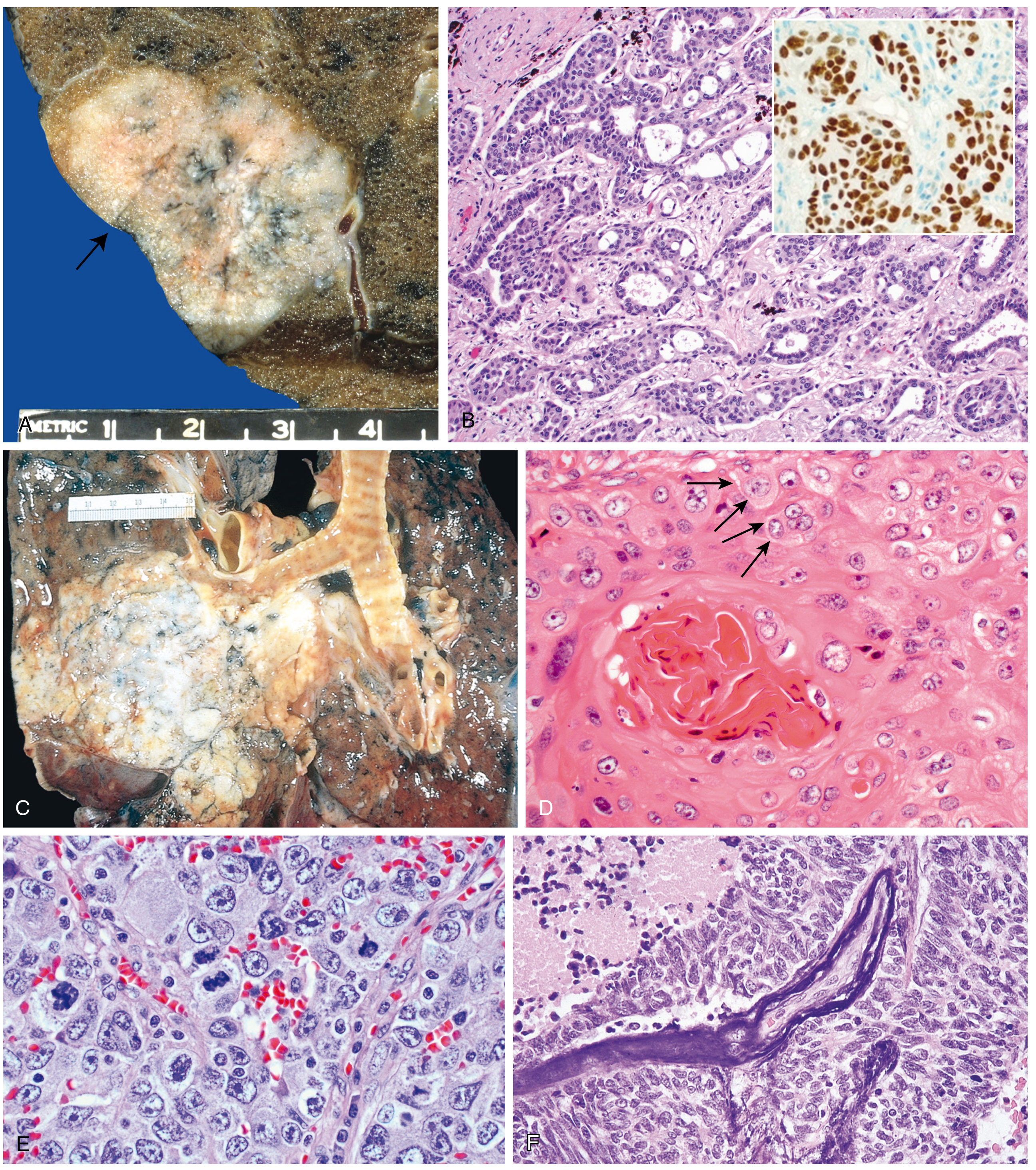
A. Adenocarcinoma (~40% - Most Common Overall)
- Most common subtype overall; the dominant type in women, never-smokers, and patients <45 years
- Location: Typically peripheral (subpleural); often associated with central scarring
- Gross: Peripheral gray-white nodule, often with pleural puckering; may be associated with areas of fibrosis
- Histology:
- Glandular differentiation or mucin production
- Patterns: acinar, papillary, micropapillary, solid, or lepidic (growth along pre-existing alveolar structures without stromal invasion)
- Adenocarcinoma in situ (AIS): tumors ≤3 cm with pure lepidic growth, no stromal invasion (the only non-invasive adenocarcinoma pattern)
- TTF-1 (thyroid transcription factor-1) positive - key IHC marker
- Precursor lesion: Atypical adenomatous hyperplasia → adenocarcinoma in situ
- Key molecular features: EGFR mutations (~20%), KRAS mutations (~30%), ALK fusions (4-6%), ROS1, HER2, MET - all targetable
B. Squamous Cell Carcinoma (~25-30%)
- Strongly associated with heavy cigarette smoking
- Location: Central (hilar/peribronchial); arises from the bronchial epithelium
- Gross: Central mass invading contiguous parenchyma; may cause obstructive atelectasis/pneumonia distal to the tumor
- Histology:
- Keratinization, keratin pearls, and intercellular bridges (key features)
- Well-differentiated tumors form obvious keratin pearls; less differentiated show sheets of cells
- Cytology of sputum shows orange-staining, keratinized cells with hyperchromatic nuclei
- Precursor lesion: Squamous dysplasia → squamous cell carcinoma in situ
- Paraneoplastic: Produces PTH-related peptide (PTHrP) → hypercalcemia
C. Small Cell Carcinoma (~15%)
- Most aggressive lung cancer; almost universally metastatic at presentation
- Strongly related to smoking (virtually 100%)
- A neuroendocrine carcinoma - derived from neuroendocrine (Kulchitsky) cells of the bronchial epithelium
- Location: Central (perihilar)
- Gross: Soft, fleshy, pale-gray central mass with extensive necrosis
- Histology:
- Small cells with scant cytoplasm, ill-defined borders
- Finely granular ("salt-and-pepper") nuclear chromatin; absent/inconspicuous nucleoli
- High mitotic rate; extensive necrosis
- Azzopardi effect: basophilic encrustation of vascular walls by DNA from necrotic tumor cells
- IHC markers: CD56, NCAM, synaptophysin, chromogranin, INSM1 (neuroendocrine markers)
- Paraneoplastic syndromes (most common with SCLC):
- ACTH → Cushing's syndrome
- ADH → SIADH
- Gastrin-releasing peptide, calcitonin
- Lambert-Eaton myasthenic syndrome (antibodies vs. presynaptic Ca²⁺ channels)
- Treatment: Chemotherapy + radiotherapy; but invariably recurs; median survival ~1 year; 5-year survival only ~5%
D. Large Cell Carcinoma (<10%)
- A diagnosis of exclusion - lacks features of adenocarcinoma, squamous cell carcinoma, or small cell carcinoma
- Location: Often peripheral
- Histology: Sheets of large cells with abundant cytoplasm, prominent nucleoli, no glandular or squamous differentiation; may have giant cells or spindle cells
- Large cell neuroendocrine carcinoma (LCNEC): a distinct aggressive subtype with neuroendocrine morphology but large-cell cytology; IHC positive for neuroendocrine markers
3. ETIOPATHOGENESIS
3.1 Etiological Factors
A. Tobacco Smoking (Primary Cause)
- ~90% of lung cancers occur in current or recent smokers
- Persons who smoke have a 10-fold or greater increased risk vs. never-smokers
- 60-fold higher risk in heavy smokers (2 packs/day for 20 years)
- Nearly linear correlation between pack-years and lung cancer frequency
- Smoking increases risk of all major types - squamous and small cell have the strongest association; adenocarcinoma is also related but is also the dominant type in never-smokers
- Risk decreases with cessation but never returns to baseline (pre-cancer genetic changes persist)
- Environmental tobacco smoke (ETS/second-hand smoke): ~20-30% increased risk in never-smokers married to long-term smokers
- Women are more susceptible to tobacco carcinogens than men (for unclear reasons)
- A large-scale genomic study found one genetic mutation per 15 cigarettes smoked
- ~65% of new US diagnoses are in former or never smokers - reflecting the changing demographics
B. Occupational and Environmental Carcinogens
| Carcinogen | Tumor Type | Interaction with Smoking |
|---|---|---|
| Asbestos | Adenocarcinoma (also mesothelioma) | 5× risk alone; ~55× with heavy smoking (synergistic) |
| Radon gas (uranium mines) | All types | Additive/synergistic |
| Arsenic, chromium, nickel | All types | Additive |
| Vinyl chloride | Sarcomatoid carcinoma | - |
| Silica | All types | Increased risk in silicosis |
| Polycyclic aromatic hydrocarbons | Squamous, small cell | - |
C. Genetic Susceptibility
- P-450 monooxygenase polymorphisms: Individuals with increased capacity to activate procarcinogens from tobacco smoke are at higher risk
- Individuals whose lymphocytes show chromosomal breakage after tobacco carcinogen exposure have increased susceptibility
- GWAS (genome-wide association studies) have identified variants in inflammation and repair pathways
- Most mutations are somatic (caused by carcinogens), but familial risk is real
D. Other Factors
- Prior lung disease (COPD, pulmonary fibrosis) increases risk
- Air pollution (PM2.5, diesel exhaust)
- HIV infection (lung cancer risk increased)
- Lung transplant recipients (especially single-lung transplant for COPD/fibrosis with prior smoking)
3.2 Pathogenesis: Molecular Steps
Lung carcinoma arises through stepwise accumulation of driver mutations that produce the hallmarks of cancer. The molecular changes follow a specific order that parallels the histologic progression:
The General Pathway:
Normal bronchial epithelium
↓ (Carcinogen exposure + "field effect")
Mutagenized mucosa — 3p deletion (early, universal)
↓
Dysplasia / atypical adenomatous hyperplasia
↓ (Additional mutations)
Carcinoma in situ — TP53 mutation, KRAS mutation (relatively late)
↓
Invasive carcinoma
↓
Metastatic disease
Key Molecular Events:
1. Chromosome 3p deletion (early, universal)
- Loss of putative tumor suppressor genes on the short arm of chromosome 3 (3p)
- Found even in benign bronchial epithelium of smokers without cancer → "field effect" (large areas of respiratory mucosa are mutagenized)
- Present in ~90% of SCLC; ~80% of NSCLC
2. TP53 mutation (late event)
- Present in ~90% of SCLC; ~50% of NSCLC
- Loss of cell cycle checkpoint → uncontrolled proliferation
3. RB (Retinoblastoma) gene mutation
- ~90% in SCLC; ~20% in NSCLC
- Loss of G1 checkpoint control
4. KRAS oncogene mutation (~30% of adenocarcinomas)
- Constitutively active RAS → continuous proliferative signaling
- Mutually exclusive with EGFR mutations (KRAS is downstream of EGFR)
- Specific KRAS variants now targetable with new drugs (e.g., sotorasib for KRAS G12C)
5. EGFR mutation (~20% of adenocarcinomas)
- Especially in non-smoking women and East Asian patients
- Activates RAS, PI3K, and other pro-growth pathways
- Targetable: gefitinib, erlotinib, osimertinib (EGFR tyrosine kinase inhibitors)
- Tumors sensitive but develop resistance
6. ALK fusion (4-6% of adenocarcinomas)
- EML4-ALK chromosomal rearrangement
- Common in non-smokers, young patients, signet-ring cell morphology
- Targetable: crizotinib, alectinib
7. ROS1, HER2, MET mutations (4-6% overall)
- Each targeted by specific drugs
8. p16/CDKN2A inactivation
- ~10% SCLC; ~50% NSCLC
- Loss of CDK4/6 inhibition → unrestrained cell cycle progression
Summary table of molecular genetics:
| Molecular Event | SCLC | NSCLC (Adeno) |
|---|---|---|
| 3p deletions | ~90% | ~80% |
| TP53 mutations | ~90% | ~50% |
| RB mutations | ~90% | ~20% |
| p16/CDKN2A | ~10% | ~50% |
| KRAS mutations | Rare | ~30% |
| EGFR mutations | Absent | ~20% |
| ALK fusions | Absent | 4-6% |
The "Field Effect" Concept:
Because carcinogens in tobacco smoke mutagenize large areas of respiratory mucosa, multiple separate foci of cancer can develop ("field cancerization"). This explains why patients who have one lung cancer cured by resection remain at high risk for developing a second primary.
3.3 Local Effects and Complications
Once a carcinoma develops, it causes local pathologic effects:
- Partial bronchial obstruction → marked focal emphysema
- Total obstruction → atelectasis
- Impaired drainage → severe suppurative bronchitis, bronchiectasis, pulmonary abscess
- Superior vena cava (SVC) syndrome: compression/invasion → venous congestion, edema of head and arms, circulatory compromise
- Pancoast tumor (apical/superior sulcus tumor): invades cervical sympathetic plexus and brachial plexus → severe pain in ulnar nerve distribution + Horner syndrome (enophthalmos, ptosis, miosis, anhidrosis)
- Pericarditis or pleural effusion: extension to pericardial/pleural sacs
- Paraneoplastic syndromes (especially SCLC): SIADH, Cushing's, hypercalcemia (squamous), Lambert-Eaton
3.4 Metastatic Routes
Lung carcinomas metastasize via:
- Lymphatics → hilar and mediastinal lymph nodes (early)
- Hematogenous → adrenal glands (most common), liver, brain, bone, kidney, and adrenal glands
- Direct spread → pleura, pericardium, great vessels, esophagus
Common sites:
- Brain (~30-40% of SCLC at presentation)
- Bone (especially SCLC)
- Liver and adrenals
- Contralateral lung
Summary Comparison Table: Major Subtypes
| Feature | Adenocarcinoma | Squamous Cell | Small Cell | Large Cell |
|---|---|---|---|---|
| Frequency | ~40% (most common) | ~25-30% | ~15% | <10% |
| Location | Peripheral | Central (hilar) | Central (hilar) | Often peripheral |
| Smoking relation | Moderate | Strong | Very strong | Moderate |
| Precursor lesion | AIS/AAH | Squamous dysplasia/CIS | None known | None known |
| Key histology | Glands, mucin, lepidic | Keratin pearls, bridges | Small cells, "oat cells", Azzopardi | Sheets of large cells |
| IHC | TTF-1, CK7 | p40, CK5/6 | CD56, synaptophysin, chromogranin | CK (nonspecific) |
| Key mutations | EGFR, KRAS, ALK | SOX2 amplification | RB, TP53, 3p del | - |
| Paraneoplastic | Rare | PTHrP → hypercalcemia | ACTH, ADH, Lambert-Eaton | Rare |
| Treatment | Surgery ± targeted/immuno | Surgery ± chemo/radiation | Chemo/radiation (NOT surgery) | Surgery ± chemo |
| Prognosis | Variable (EGFR+ better) | Moderate | Poor (median 1 yr) | Poor |
- Robbins & Kumar Basic Pathology, Chapter 11 (Lung)
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th ed., Chapter 15
- Harrison's Principles of Internal Medicine, 22nd ed., Chapter 83
This is a shared conversation. Sign in to Orris to start your own chat.