Retinal degenerations
Reading File
Finding Sources
Searching PubMed
"retinal degeneration"[MeSH Terms] AND treatment
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
I now have rich content from multiple authoritative textbooks. Let me compile the comprehensive answer.
Retinal Degenerations
Retinal degenerations are a broad group of conditions — hereditary and acquired — that result in progressive loss of photoreceptors, retinal pigment epithelium (RPE), or both, leading to varying degrees of visual impairment and blindness.
1. Age-Related Macular Degeneration (AMD)
AMD is the leading cause of gradual, painless, bilateral central visual loss in the elderly. It primarily affects the RPE of the macula, with prevalence rising sharply each decade after age 55. Approximately 8.5% of global blindness is attributable to AMD. Susceptibility is linked to variants in complement factor H (CFH) and other complement regulatory genes (implying excessive alternative complement pathway activity), as well as cigarette smoking and metabolic factors.
Dry (Non-exudative) AMD
- Begins with accumulation of extracellular deposits called drusen within the RPE basement membrane (Bruch's membrane)
- Drusen appear as discrete yellow lesions clustered in the macula; soft drusen >60 μm are significant
- Lipofuscin accumulates in stressed RPE cells
- Progresses to geographic atrophy — well-defined zones of RPE loss — with slow central visual decline (months to decades)
- Peripheral retina is spared: patients lose face recognition but retain ambulation
Fundus image — AMD (dry + wet):
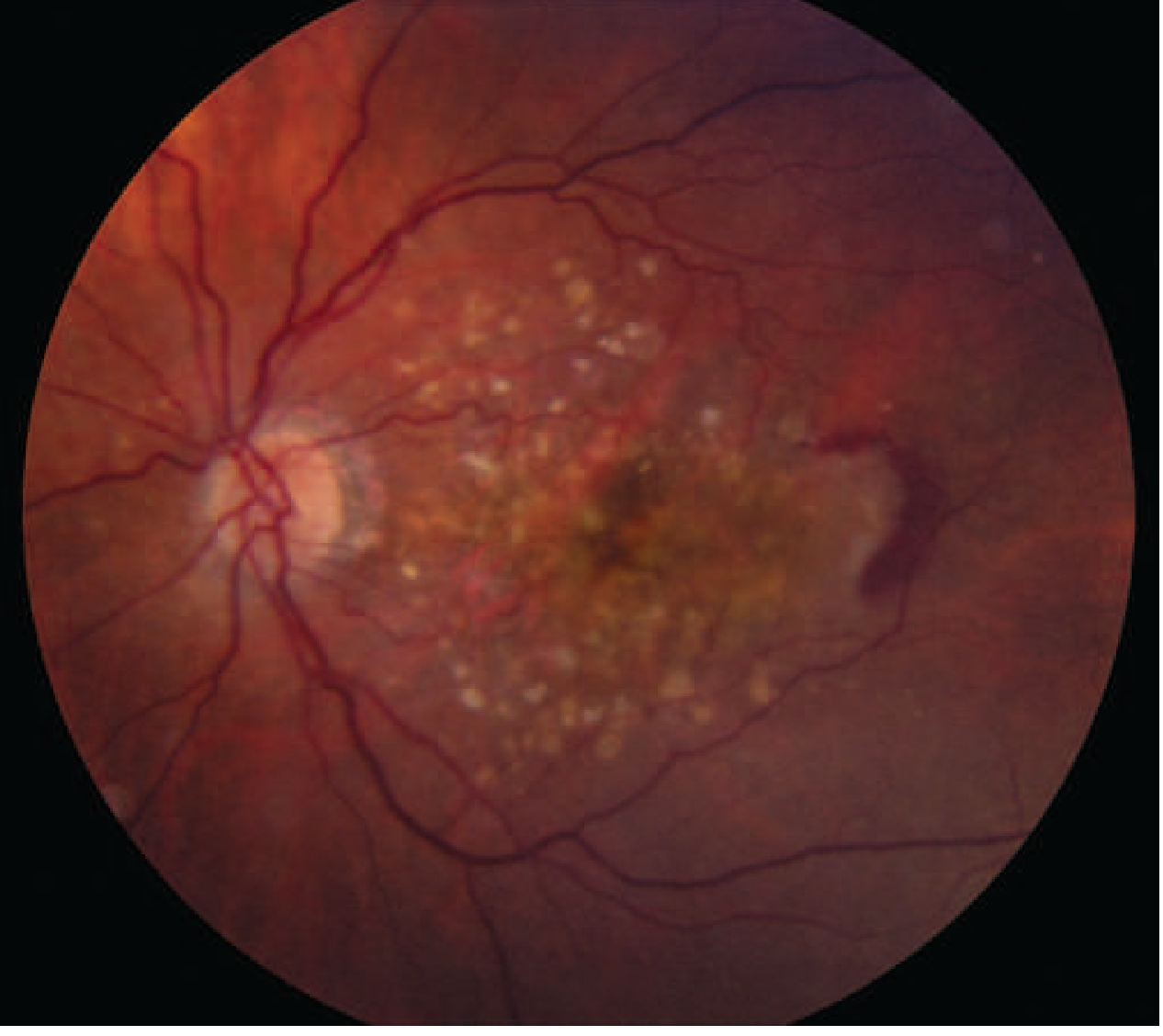
Wet (Neovascular/Exudative) AMD
- Occurs when choroidal neovascular (CNV) vessels penetrate Bruch's membrane into the subretinal space
- Leakage causes retinal elevation, metamorphopsia, and blurred vision; hemorrhage can cause sudden visual loss
- Results in disciform (round) macular scar and permanent central vision loss if untreated
- Detected by fluorescein angiography and OCT
Treatment
| Form | Management |
|---|---|
| Dry AMD | Vitamins C & E, lutein, zeaxanthin, zinc, copper (AREDS2 formula); stop smoking; control BP and glucose |
| Wet AMD | Intravitreal anti-VEGF injections (monthly initially): bevacizumab, ranibizumab, aflibercept, brolucizumab; faricimab (dual angiopoietin-2 + VEGF-A inhibitor, newer); photodynamic therapy in selected cases |
⚠️ Recent evidence (2024–2025): A network meta-analysis (PMID 39043575) confirmed faricimab's efficacy for neovascular AMD. A 2025 network meta-analysis (PMID 40241463) compared intravitreal anti-VEGF agents comprehensively. OCT biomarkers predicting progression to late AMD have been systematically reviewed (PMID 38154619).
2. Retinitis Pigmentosa (RP)
RP is the most common hereditary retinal degeneration, with a prevalence of 1:3,000–1:5,000. It is a clinically and genetically diverse group of rod-cone dystrophies characterized by progressive photoreceptor and RPE degeneration.
Genetics
- Over 100 gene loci implicated in non-syndromic RP
- Inheritance patterns: autosomal dominant (AD), autosomal recessive (AR), X-linked, or sporadic
- Most AD-RP: mutations in rhodopsin gene (phototransduction cascade)
- Most X-linked RP (90%): mutations in RPGR gene (connecting cilium protein)
- Pathways affected: phototransduction cascade, retinoid cycle, photoreceptor structure, RPE function
- ~50% of affected individuals have no identified molecular defect
- Cell death mechanism: apoptosis, possibly from accumulation of misfolded proteins
Symptoms
- Nyctalopia (night blindness) — earliest feature; rods affected first
- Progressive peripheral visual field constriction with ring scotoma
- Photopsia
- Central visual acuity loss is a later feature (unless cataract develops earlier)
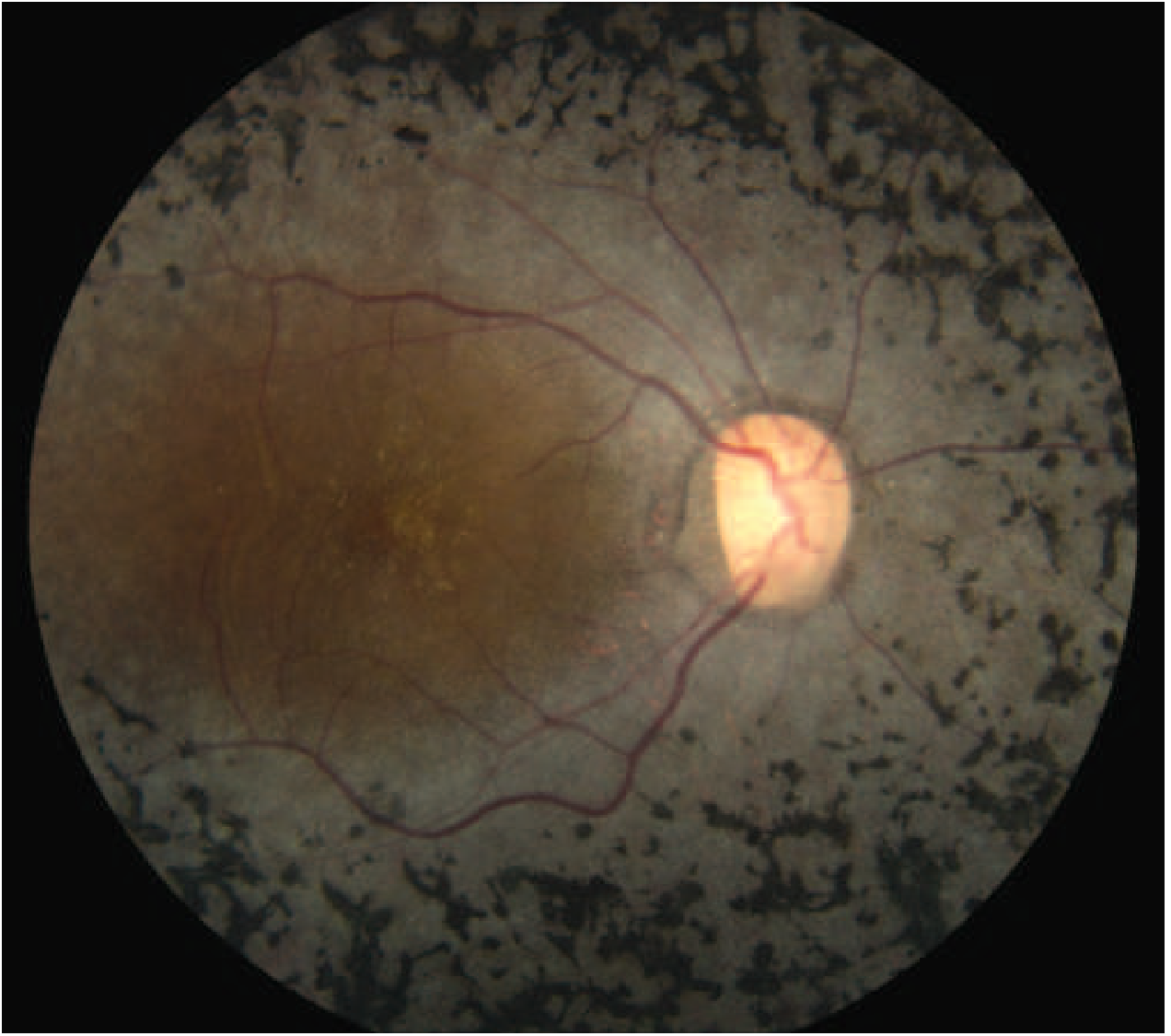
Classic Triad on Fundoscopy
- Bone-spicule pigmentation — mid-peripheral intraretinal perivascular pigment deposits
- Arteriolar attenuation
- "Waxy" disc pallor (optic nerve head atrophy)
Fundus image — Advanced RP:
Investigations
- ERG: reduced or absent scotopic (rod) responses — extinguished in advanced disease; hallmark test
- Fundus autofluorescence (FAF): posterior pole hyperautofluorescent rings
- Visual field testing: peripheral constriction
- OCT: macular changes (CMO, ERM, atrophy)
- Genetic panel testing
Management
- No curative treatment for most forms
- Cystoid macular oedema (CMO): oral acetazolamide or topical carbonic anhydrase inhibitors
- Light protection: amber spectacles (blocking <550 nm) may reduce glare and improve contrast
- Dietary: vitamin A supplementation (effect uncertain, gene-dependent; caution with toxicity)
- Avoid retinotoxic drugs: isotretinoin, phenothiazines, hydroxychloroquine, tamoxifen, vigabatrin
- Gene therapy: Leber congenital amaurosis (LCA) with RPE65 mutations — voretigene neparvovec (Luxturna) approved, with improvement in visual function
- Cataract surgery beneficial when lens opacity limits vision
Syndromic RP (20–30% of cases)
RP occurs as part of systemic syndromes — usually AR or mitochondrial inheritance:
| Syndrome | Features |
|---|---|
| Usher syndrome | RP + sensorineural hearing loss ± vestibular dysfunction (most common cause of deaf-blindness) |
| Bardet-Biedl syndrome | RP + obesity, polydactyly, hypogonadism, renal anomalies, intellectual disability |
| Kearns-Sayre syndrome (mitochondrial) | RP + progressive external ophthalmoplegia, heart block |
| Refsum disease | RP + polyneuropathy, ataxia, hearing loss, elevated phytanic acid |
| Bassen-Kornzweig disease (abetalipoproteinemia) | RP + fat malabsorption, acanthocytosis |
| Olivopontocerebellar degeneration | RP + cerebellar ataxia |
| NARP syndrome | Neuropathy, ataxia, RP (mitochondrial) |
3. Other Hereditary Retinal Dystrophies
Leber Congenital Amaurosis (LCA)
- Severe early-onset retinal dystrophy (~5% of RP); presents at birth or early infancy
- Mutations in RPE65, CEP290, CRB1, GUCY2D, and others
- Gene therapy (voretigene neparvovec) for RPE65-associated LCA is an approved treatment
Stargardt Disease (Fundus Flavimaculatus)
- Most common hereditary macular dystrophy in children/young adults; onset usually <25 years
- AR inheritance; ABCA4 gene mutations
- Features: central visual loss, yellow-white flecks in the posterior pole, bull's-eye maculopathy
- ERG usually normal initially
- No established treatment; avoid excess vitamin A (substrate for toxic bisretinoids)
Cone and Cone-Rod Dystrophies
- Primary cone degeneration (vs. RP's primary rod degeneration)
- Symptoms: photophobia, colour vision loss, central scotoma early; ERG shows absent photopic responses
- May progress to rod involvement (cone-rod dystrophy)
Best Vitelliform Macular Dystrophy
- Second most common macular dystrophy (after Stargardt)
- BEST1 (bestrophin) gene mutations on chromosome 11q13
- Characteristic "egg yolk" macular lesion on fundoscopy
Toxic / Drug-Induced Retinopathy
Resembles RP and must be excluded:
- Chloroquine/hydroxychloroquine: bull's-eye maculopathy; screening with OCT and visual fields required for long-term users
- Thioridazine (phenothiazine): diffuse retinal pigmentary changes
- Vigabatrin: peripheral visual field constriction
Summary: Distinguishing Features
| Condition | Primary Deficit | Visual Loss Type | Key Finding |
|---|---|---|---|
| Dry AMD | RPE/photoreceptors (macula) | Central, gradual | Drusen, geographic atrophy |
| Wet AMD | Choroidal neovascularization | Central, sudden-on-gradual | CNV, subretinal fluid/haem |
| RP | Rod > cone (periphery→centre) | Peripheral → central | Bone spicules, waxy disc |
| Stargardt | Cone (macula) | Central, early onset | Flecks, bull's-eye |
| LCA | Rods + cones (severe, congenital) | Severe from birth | Nystagmus, absent ERG |
| Cone dystrophy | Cone | Central + colour | Photopic ERG absent |
Sources: Harrison's Principles of Internal Medicine 22E (2025); Kanski's Clinical Ophthalmology 10th ed.; Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine; The Wills Eye Manual.
This is a shared conversation. Sign in to Orris to start your own chat.