Give me proper explanation why HHH syndrome occurs. Mechanism about it’s biochemistry and treatment plan
HHH Syndrome (Hyperornithinemia–Hyperammonemia–Homocitrullinuria)
The Normal Urea Cycle — Context First
The Defect — ORC1 (Ornithine/Citrulline Carrier)
- Imports cytosolic ornithine → mitochondria
- Exports newly synthesized citrulline → cytosol
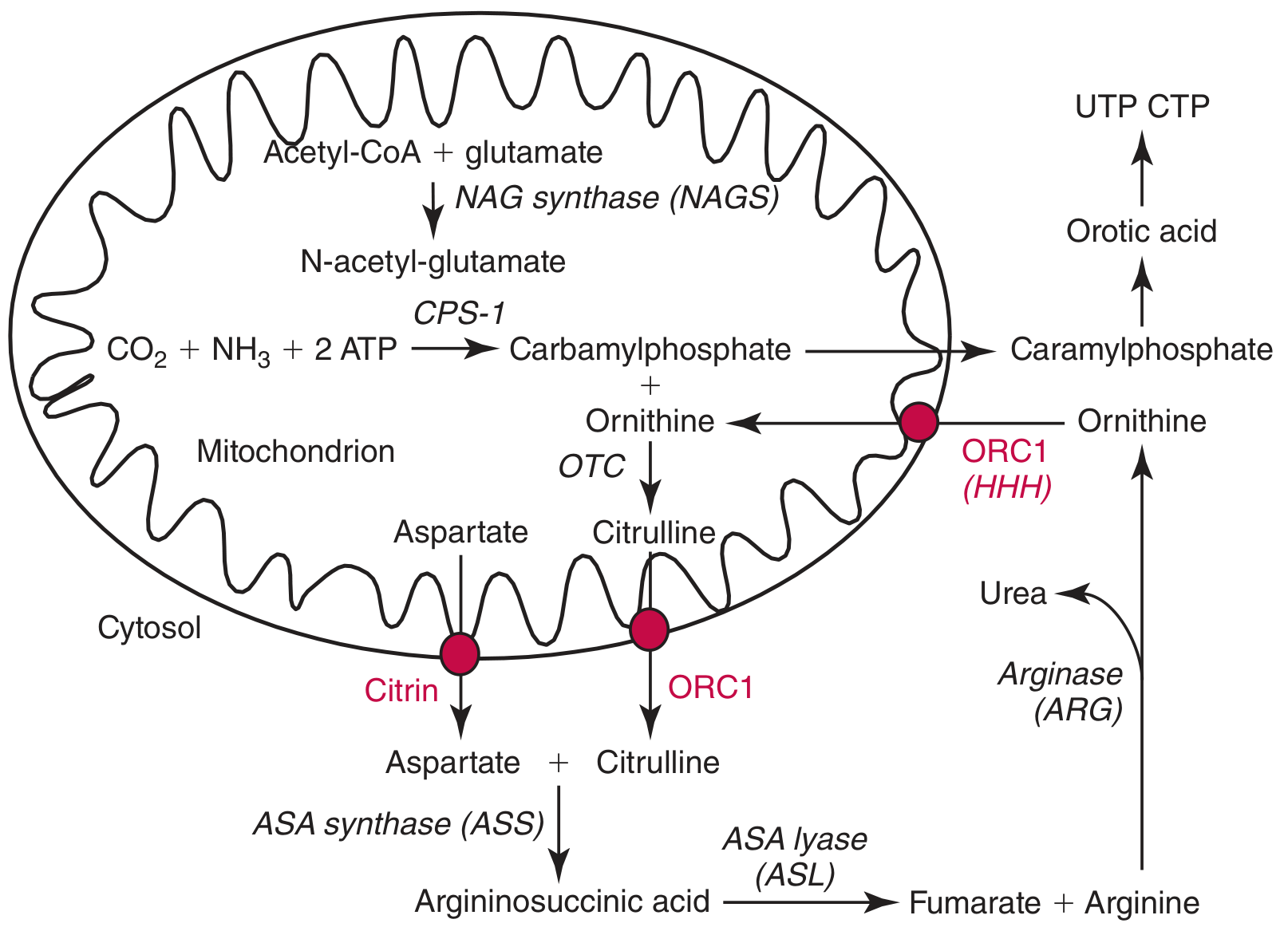
Step-by-Step Biochemical Mechanism
1. Ornithine Cannot Enter the Mitochondrion
- ORC1 is non-functional → ornithine accumulates in the cytosol
- This produces hyperornithinemia
2. Urea Cycle Comes to a Halt
- Without intramitochondrial ornithine, OTC has no substrate
- Carbamoyl phosphate (CP) accumulates inside the mitochondria
- The urea cycle is functionally blocked → hyperammonemia (NH₃ cannot be converted to urea)
3. Carbamoyl Phosphate Is Diverted to Lysine
- With no ornithine to accept it, the accumulated mitochondrial carbamoyl phosphate reacts with lysine (a dibasic amino acid), via an alternative reaction
- This produces homocitrulline (the lysine analogue of citrulline)
- Homocitrulline is excreted in urine → homocitrullinuria
4. Secondary Brain Toxicity
- NH₃ cannot be safely converted to urea
- The brain attempts to detoxify NH₃ by converting glutamate → glutamine (via glutamine synthetase)
- Glutamine and glutamate accumulate in astrocytes → osmotic brain edema
- This is the primary mechanism of hyperammonemic encephalopathy
- Elevated NH₃ also stimulates hyperventilation → respiratory alkalosis
Summary Table of the Three Hallmarks
| Feature | Mechanism |
|---|---|
| Hyperornithinemia | Ornithine cannot enter mitochondria → cytosolic accumulation |
| Hyperammonemia | Urea cycle blocked (no ornithine substrate for OTC) → NH₃ accumulates |
| Homocitrullinuria | Carbamoyl phosphate reacts with lysine (substitute for ornithine) → homocitrulline excreted |
Clinical Features
- Neonatal/early onset: lethargy, poor feeding, vomiting, progressive encephalopathy, coma, apnea (from severe hyperammonemia)
- Late/partial onset (more common): protein intolerance, episodic vomiting, behavioral problems, developmental delay, intellectual disability, recurrent ataxia, seizures
- Symptoms are triggered or worsened by high-protein intake or catabolic stress (fasting, illness)
- Progressive spastic paraparesis is a recognized late complication
- Episodes mimic GI disorders, food allergies, or behavioral problems → frequent misdiagnosis
Laboratory Diagnosis
| Finding | Value |
|---|---|
| Plasma ammonia | Elevated (episodic or persistent) |
| Plasma ornithine | Elevated |
| Urine homocitrulline | Elevated |
| Plasma citrulline | Low-normal (urea cycle is blocked upstream of citrulline synthesis) |
| Plasma glutamine & alanine | Elevated (nonspecific to all urea cycle disorders) |
| Urine orotic acid | Normal (unlike OTC deficiency) |
Treatment Plan
Acute Crisis Management
- Stop protein intake temporarily to reduce nitrogen load
- IV glucose + lipids to reverse catabolism (insulin may be used)
- Nitrogen scavengers:
- Sodium benzoate — conjugates glycine → hippurate (excreted)
- Sodium phenylacetate or phenylbutyrate — conjugates glutamine → phenylacetylglutamine (excreted)
- These bypass the urea cycle for nitrogen excretion
- Arginine supplementation — provides downstream urea cycle substrate; supports protein synthesis resumption
- Hemodialysis — if ammonia does not fall rapidly within hours despite above measures
Long-Term Chronic Management
| Intervention | Rationale |
|---|---|
| Low-protein diet (frequent small meals) | Reduces nitrogen load; avoids ammonia spikes |
| Ornithine supplementation | Provides the missing intramitochondrial substrate; improves urea cycle flux — directly addresses the block |
| Citrulline supplementation | Used in proximal urea cycle defects (OTC/CPS-1); also useful here |
| Ammonia scavengers (phenylbutyrate, benzoate, glycerol phenylbutyrate — oral) | Long-term nitrogen excretion bypass |
| Liver transplantation | Curative for enzymatic/transporter defects restricted to liver; restores normal ORC1 function |
Ornithine administration is notable in HHH specifically: by supplying exogenous ornithine that partially compensates for impaired import, it can improve urea cycle function and reduce ammonia levels. — Bradley and Daroff's Neurology in Clinical Practice
Inheritance and Genetics
- Autosomal recessive — both copies of SLC25A15 must be mutated
- Gene encodes a mitochondrial inner membrane solute carrier
- Considerable phenotypic variability even among patients with the same mutation
Make a pdf out of above content after adding a clinical case presenting HHH syndrome so that it can be presented in class
ls -la /home/daytona/workspace/ 2>/dev/null || echo "workspace empty or missing"
mkdir -p /home/daytona/workspace/hhh-syndrome
~/hhh-syndrome/generate_pdf.py
import json, subprocess, base64, os
from reportlab.lib.pagesizes import A4
from reportlab.lib import colors
from reportlab.lib.units import cm
from reportlab.lib.styles import getSampleStyleSheet, ParagraphStyle
from reportlab.lib.enums import TA_LEFT, TA_CENTER, TA_JUSTIFY
from reportlab.platypus import (
SimpleDocTemplate, Paragraph, Spacer, Table, TableStyle,
HRFlowable, PageBreak, Image, KeepTogether
)
from reportlab.platypus.flowables import HRFlowable
from reportlab.lib.colors import HexColor
OUTPUT = "/home/daytona/workspace/hhh-syndrome/HHH_Syndrome.pdf"
# ── Colours ──────────────────────────────────────────────────────────────────
DARK_BLUE = HexColor("#1a3a5c")
MED_BLUE = HexColor("#2e6da4")
LIGHT_BLUE = HexColor("#dce9f5")
ACCENT = HexColor("#c0392b")
CASE_BG = HexColor("#fdf3e7")
CASE_BORDER = HexColor("#e67e22")
TABLE_HDR = HexColor("#2e6da4")
TABLE_ALT = HexColor("#eaf2fb")
GRAY_LINE = HexColor("#cccccc")
WHITE = colors.white
BLACK = colors.black
# ── Document ─────────────────────────────────────────────────────────────────
doc = SimpleDocTemplate(
OUTPUT,
pagesize=A4,
leftMargin=2*cm, rightMargin=2*cm,
topMargin=2.5*cm, bottomMargin=2.5*cm,
title="HHH Syndrome – Biochemistry & Clinical Overview",
author="Medical Education"
)
W, H = A4
CONTENT_W = W - 4*cm
# ── Styles ────────────────────────────────────────────────────────────────────
base = getSampleStyleSheet()
def style(name, parent="Normal", **kw):
return ParagraphStyle(name, parent=base[parent], **kw)
S = {
"title": style("s_title", "Title",
fontSize=26, textColor=WHITE,
spaceAfter=4, leading=32, alignment=TA_CENTER),
"subtitle": style("s_sub", "Normal",
fontSize=12, textColor=LIGHT_BLUE,
alignment=TA_CENTER, spaceAfter=2),
"omim": style("s_omim", "Normal",
fontSize=10, textColor=LIGHT_BLUE,
alignment=TA_CENTER, spaceAfter=0),
"h1": style("s_h1", "Heading1",
fontSize=14, textColor=WHITE,
spaceBefore=14, spaceAfter=6, leading=18),
"h2": style("s_h2", "Heading2",
fontSize=11, textColor=DARK_BLUE,
spaceBefore=10, spaceAfter=4, leading=14),
"body": style("s_body", "Normal",
fontSize=10, leading=15, spaceAfter=6,
alignment=TA_JUSTIFY),
"bullet": style("s_bullet", "Normal",
fontSize=10, leading=14, spaceAfter=3,
leftIndent=16, bulletIndent=4),
"caption": style("s_cap", "Normal",
fontSize=8, textColor=colors.grey,
alignment=TA_CENTER, spaceAfter=4),
"case_title": style("s_ctitle", "Normal",
fontSize=13, textColor=CASE_BORDER,
fontName="Helvetica-Bold", spaceAfter=6),
"case_body": style("s_cbody", "Normal",
fontSize=10, leading=15, spaceAfter=5,
alignment=TA_JUSTIFY),
"case_q": style("s_cq", "Normal",
fontSize=10, leading=14, spaceAfter=4,
fontName="Helvetica-Bold", textColor=DARK_BLUE),
"case_a": style("s_ca", "Normal",
fontSize=10, leading=14, spaceAfter=6,
leftIndent=12, textColor=HexColor("#2c3e50")),
"ref": style("s_ref", "Normal",
fontSize=8, textColor=colors.grey,
leading=12, spaceAfter=2),
"tbl_hdr": style("s_thdhr", "Normal",
fontSize=9.5, textColor=WHITE,
fontName="Helvetica-Bold", alignment=TA_CENTER),
"tbl_cell": style("s_tcell", "Normal",
fontSize=9.5, leading=13, alignment=TA_LEFT),
"tbl_cell_c": style("s_tcellc", "Normal",
fontSize=9.5, leading=13, alignment=TA_CENTER),
}
# ── Helpers ───────────────────────────────────────────────────────────────────
def section_header(text):
"""Blue banner heading."""
tbl = Table([[Paragraph(text, S["h1"])]], colWidths=[CONTENT_W])
tbl.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), MED_BLUE),
("ROUNDEDCORNERS", [4]),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING",(0,0),(-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 10),
]))
return tbl
def bullet(text, symbol="•"):
return Paragraph(f"<bullet>{symbol}</bullet> {text}", S["bullet"])
def hr(color=GRAY_LINE, thickness=0.5):
return HRFlowable(width="100%", thickness=thickness, color=color, spaceAfter=4, spaceBefore=4)
def two_col_table(rows, col_headers, col_widths, alt=True):
"""Generic styled table."""
data = [[Paragraph(h, S["tbl_hdr"]) for h in col_headers]] + \
[[Paragraph(str(c), S["tbl_cell"]) for c in row] for row in rows]
style_cmds = [
("BACKGROUND", (0,0), (-1,0), TABLE_HDR),
("ROWBACKGROUNDS",(0,1), (-1,-1), [WHITE, TABLE_ALT] if alt else [WHITE]),
("GRID", (0,0), (-1,-1), 0.4, GRAY_LINE),
("VALIGN", (0,0), (-1,-1), "TOP"),
("TOPPADDING", (0,0), (-1,-1), 5),
("BOTTOMPADDING", (0,0), (-1,-1), 5),
("LEFTPADDING", (0,0), (-1,-1), 7),
("RIGHTPADDING", (0,0), (-1,-1), 7),
]
t = Table(data, colWidths=col_widths)
t.setStyle(TableStyle(style_cmds))
return t
def case_box(flowables):
"""Orange-bordered clinical case box."""
inner = Table([[f] for f in flowables], colWidths=[CONTENT_W - 1.6*cm])
inner.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), CASE_BG),
("LEFTPADDING", (0,0), (-1,-1), 10),
("RIGHTPADDING", (0,0), (-1,-1), 10),
("TOPPADDING", (0,0), (-1,-1), 4),
("BOTTOMPADDING",(0,0), (-1,-1), 4),
]))
outer = Table([[inner]], colWidths=[CONTENT_W])
outer.setStyle(TableStyle([
("BOX", (0,0), (-1,-1), 2, CASE_BORDER),
("ROUNDEDCORNERS", [6]),
("TOPPADDING", (0,0), (-1,-1), 0),
("BOTTOMPADDING",(0,0), (-1,-1), 0),
("LEFTPADDING", (0,0), (-1,-1), 0),
("RIGHTPADDING", (0,0), (-1,-1), 0),
]))
return outer
# ── Download urea-cycle diagram ───────────────────────────────────────────────
IMG_URL = "https://cdn.orris.care/cdss_images/0c9e9fa9f44799ed524b40e6c5b281e4e2c20fba4445ac19cbb5b2f8ff34ccfe.png"
IMG_PATH = "/home/daytona/workspace/hhh-syndrome/urea_cycle.png"
if not os.path.exists(IMG_PATH):
result = json.loads(subprocess.check_output([
"node", "/home/daytona/skills/shared/scripts/fetch_images.js", IMG_URL
]))
if result and result[0].get("base64"):
raw = base64.b64decode(result[0]["base64"].split(",")[1])
with open(IMG_PATH, "wb") as f:
f.write(raw)
print("Image downloaded OK")
else:
print("Image download failed:", result)
IMG_PATH = None
else:
print("Image already exists")
# ══════════════════════════════════════════════════════════════════════════════
# STORY
# ══════════════════════════════════════════════════════════════════════════════
story = []
# ── COVER BANNER ─────────────────────────────────────────────────────────────
cover_inner = [
Spacer(1, 0.5*cm),
Paragraph("HHH Syndrome", S["title"]),
Paragraph("Hyperornithinemia · Hyperammonemia · Homocitrullinuria", S["subtitle"]),
Paragraph("OMIM #238970 | Autosomal Recessive | Gene: SLC25A15 (ORC1)", S["omim"]),
Spacer(1, 0.5*cm),
]
cover_tbl = Table([[f] for f in cover_inner], colWidths=[CONTENT_W])
cover_tbl.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), DARK_BLUE),
("ROUNDEDCORNERS", [8]),
("LEFTPADDING", (0,0), (-1,-1), 16),
("RIGHTPADDING", (0,0), (-1,-1), 16),
("TOPPADDING", (0,0), (-1,-1), 2),
("BOTTOMPADDING",(0,0), (-1,-1), 2),
]))
story.append(cover_tbl)
story.append(Spacer(1, 0.5*cm))
# ── SECTION 1: NORMAL UREA CYCLE ─────────────────────────────────────────────
story.append(section_header("1. The Normal Urea Cycle — Context"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph(
"The urea cycle is the liver's primary pathway for converting toxic ammonia (NH<sub>3</sub>) — "
"derived from amino acid catabolism — into urea for urinary excretion. The cycle spans two "
"cellular compartments: the <b>mitochondrial matrix</b> and the <b>cytosol</b>.",
S["body"]
))
story.append(Paragraph(
"A critical bottleneck is the <b>import of cytosolic ornithine into the mitochondria</b>, "
"where it accepts carbamoyl phosphate (synthesised by CPS-1) via ornithine transcarbamylase "
"(OTC) to produce <b>citrulline</b>. Citrulline is then exported back to the cytosol to "
"continue the cycle and ultimately generate urea. Both ornithine import and citrulline "
"export are mediated by the same transporter: <b>ORC1</b>.",
S["body"]
))
# Urea cycle diagram
if IMG_PATH and os.path.exists(IMG_PATH):
img = Image(IMG_PATH, width=12*cm, height=9*cm)
img_tbl = Table([[img]], colWidths=[CONTENT_W])
img_tbl.setStyle(TableStyle([
("ALIGN", (0,0), (-1,-1), "CENTER"),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING",(0,0), (-1,-1), 2),
]))
story.append(img_tbl)
story.append(Paragraph(
"Figure 1. The urea cycle. The ORC1 transporter (highlighted in red) is the site of the "
"defect in HHH syndrome. ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; "
"ARG, arginase; CPS-1, carbamyl phosphate synthase 1; OTC, ornithine transcarbamylase.",
S["caption"]
))
story.append(Spacer(1, 0.3*cm))
# ── SECTION 2: THE DEFECT ─────────────────────────────────────────────────────
story.append(section_header("2. The Primary Defect — ORC1 (SLC25A15)"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph(
"HHH syndrome is caused by <b>loss-of-function mutations in the <i>SLC25A15</i> gene</b>, "
"which encodes <b>ORC1</b> — the mitochondrial inner membrane ornithine/citrulline antiporter. "
"ORC1 normally performs two coupled functions simultaneously:",
S["body"]
))
story.append(bullet("Imports cytosolic <b>ornithine</b> → mitochondrial matrix"))
story.append(bullet("Exports newly synthesised <b>citrulline</b> → cytosol"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph(
"When ORC1 is non-functional, both transport steps fail, triggering a cascade of three "
"simultaneous metabolic consequences.",
S["body"]
))
# ── SECTION 3: BIOCHEMICAL MECHANISM ─────────────────────────────────────────
story.append(section_header("3. Step-by-Step Biochemical Mechanism"))
story.append(Spacer(1, 0.2*cm))
mech_steps = [
("Step 1", "Ornithine accumulates in cytosol",
"Cytosolic ornithine cannot cross the inner mitochondrial membrane. "
"It builds up in the cytosol → <b>HYPERORNITHINEMIA</b>."),
("Step 2", "Urea cycle is blocked",
"Without intramitochondrial ornithine, OTC has no substrate. "
"Carbamoyl phosphate (CP) accumulates inside mitochondria. "
"Ammonia cannot be incorporated into the cycle → <b>HYPERAMMONEMIA</b>."),
("Step 3", "CP reacts with lysine",
"With no ornithine to accept it, excess mitochondrial carbamoyl phosphate "
"reacts with <b>lysine</b> (a dibasic amino acid) via an alternative, low-specificity reaction. "
"This produces <b>homocitrulline</b> (the lysine analogue of citrulline), "
"which is excreted in urine → <b>HOMOCITRULLINURIA</b>."),
("Step 4", "Brain toxicity",
"Elevated NH<sub>3</sub> cannot be converted to urea. The brain detoxifies it by "
"converting glutamate → <b>glutamine</b> (via glutamine synthetase). "
"Glutamine accumulates in astrocytes → <b>osmotic cerebral oedema</b>. "
"Excess NH<sub>3</sub> also drives hyperventilation → <b>respiratory alkalosis</b>."),
]
for step_num, step_title, step_text in mech_steps:
row_data = [
[Paragraph(f"<b>{step_num}</b>", S["tbl_cell_c"]),
Paragraph(f"<b>{step_title}</b>", S["tbl_cell"]),
Paragraph(step_text, S["tbl_cell"])],
]
t = Table(row_data, colWidths=[1.5*cm, 4.5*cm, CONTENT_W - 6*cm])
t.setStyle(TableStyle([
("BACKGROUND", (0,0), (0,0), MED_BLUE),
("TEXTCOLOR", (0,0), (0,0), WHITE),
("BACKGROUND", (1,0), (1,0), LIGHT_BLUE),
("GRID", (0,0), (-1,-1), 0.4, GRAY_LINE),
("VALIGN", (0,0), (-1,-1), "TOP"),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING",(0,0), (-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 7),
("RIGHTPADDING", (0,0), (-1,-1), 7),
]))
story.append(t)
story.append(Spacer(1, 0.15*cm))
# ── SECTION 4: HALLMARKS TABLE ────────────────────────────────────────────────
story.append(Spacer(1, 0.2*cm))
story.append(section_header("4. Summary of the Three Hallmarks"))
story.append(Spacer(1, 0.2*cm))
hallmarks = [
["Hyperornithinemia",
"ORC1 defect → ornithine cannot enter mitochondria → cytosolic accumulation",
"Plasma ornithine elevated"],
["Hyperammonemia",
"No ornithine substrate for OTC → urea cycle blocked → NH3 accumulates",
"Plasma ammonia elevated; glutamine & alanine elevated"],
["Homocitrullinuria",
"Excess carbamoyl phosphate reacts with lysine → homocitrulline formed",
"Urine homocitrulline elevated"],
]
story.append(two_col_table(
hallmarks,
["Feature", "Mechanism", "Lab Marker"],
[3.5*cm, 7.5*cm, 4.5*cm]
))
# ── SECTION 5: CLINICAL FEATURES ──────────────────────────────────────────────
story.append(PageBreak())
story.append(section_header("5. Clinical Features"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Neonatal / Early Onset (severe enzymatic block):</b>", S["h2"]))
for f in [
"Lethargy, poor feeding, vomiting within the first days of life",
"Progressive encephalopathy → coma → apnea",
"Caused by severe acute hyperammonemia",
]:
story.append(bullet(f))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Late / Partial Onset (more common presentation):</b>", S["h2"]))
for f in [
"Protein intolerance — child self-restricts high-protein foods",
"Episodic vomiting, irritability, lethargy triggered by high protein intake or illness",
"Developmental delay, intellectual disability, learning difficulties",
"Seizures, recurrent ataxia, behavioural problems",
"Progressive <b>spastic paraparesis</b> — a recognised late complication",
"Frequently <b>misdiagnosed</b> as GI disorder, food allergy, or behavioural problem",
]:
story.append(bullet(f))
# ── SECTION 6: DIAGNOSIS ──────────────────────────────────────────────────────
story.append(Spacer(1, 0.3*cm))
story.append(section_header("6. Laboratory Diagnosis"))
story.append(Spacer(1, 0.2*cm))
diag_rows = [
["Plasma ammonia", "Elevated (episodic or persistent)"],
["Plasma ornithine", "Elevated"],
["Urine homocitrulline", "Elevated (pathognomonic)"],
["Urine orotic acid", "Normal (distinguishes from OTC deficiency)"],
["Plasma citrulline", "Low-normal (urea cycle blocked upstream)"],
["Plasma glutamine & alanine", "Elevated (non-specific to all UCDs)"],
["Plasma arginine", "Low"],
["Genetic testing", "SLC25A15 mutation analysis — confirmatory"],
]
story.append(two_col_table(
diag_rows,
["Investigation", "Finding in HHH Syndrome"],
[5.5*cm, CONTENT_W - 5.5*cm]
))
# ── SECTION 7: TREATMENT ──────────────────────────────────────────────────────
story.append(Spacer(1, 0.3*cm))
story.append(section_header("7. Treatment Plan"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>A. Acute Crisis Management</b>", S["h2"]))
acute = [
["1", "Halt protein intake",
"Temporarily stop protein to reduce nitrogen load immediately"],
["2", "IV glucose + lipids ± insulin",
"Provide calories to reverse catabolism; insulin promotes anabolism"],
["3", "Nitrogen scavengers (IV)",
"<b>Sodium benzoate</b> — conjugates glycine → hippurate (renal excretion)\n"
"<b>Sodium phenylacetate</b> — conjugates glutamine → phenylacetylglutamine"],
["4", "Arginine supplementation",
"Provides downstream urea cycle substrate; supports protein synthesis resumption"],
["5", "Hemodialysis",
"If ammonia does not fall within hours despite above measures — mandatory"],
]
t_acute = Table(
[[Paragraph(f"<b>{r[0]}</b>", S["tbl_cell_c"]),
Paragraph(f"<b>{r[1]}</b>", S["tbl_cell"]),
Paragraph(r[2], S["tbl_cell"])] for r in acute],
colWidths=[1.2*cm, 4.8*cm, CONTENT_W - 6*cm]
)
t_acute.setStyle(TableStyle([
("BACKGROUND", (0,0), (0,-1), MED_BLUE),
("TEXTCOLOR", (0,0), (0,-1), WHITE),
("ROWBACKGROUNDS",(1,0),(2,-1), [WHITE, TABLE_ALT]),
("GRID", (0,0), (-1,-1), 0.4, GRAY_LINE),
("VALIGN", (0,0), (-1,-1), "TOP"),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING",(0,0), (-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 7),
("RIGHTPADDING", (0,0), (-1,-1), 7),
]))
story.append(t_acute)
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("<b>B. Long-Term Chronic Management</b>", S["h2"]))
chronic_rows = [
["Low-protein diet (frequent small meals)",
"Reduces nitrogen load; prevents ammonia spikes"],
["Ornithine supplementation",
"Directly addresses the block — exogenous ornithine partially compensates "
"for impaired mitochondrial import, improving urea cycle flux"],
["Citrulline supplementation",
"Provides downstream substrate, especially useful in proximal cycle blocks"],
["Oral ammonia scavengers\n(phenylbutyrate, benzoate,\nglycerol phenylbutyrate)",
"Long-term nitrogen excretion bypass around the blocked cycle"],
["Liver transplantation",
"Curative — restores ORC1 function in hepatocytes, normalises the urea cycle"],
]
story.append(two_col_table(
chronic_rows,
["Intervention", "Rationale"],
[5.5*cm, CONTENT_W - 5.5*cm]
))
# ── SECTION 8: CLINICAL CASE ──────────────────────────────────────────────────
story.append(PageBreak())
story.append(section_header("8. Clinical Case Presentation"))
story.append(Spacer(1, 0.3*cm))
case_content = [
Paragraph("CASE: The Child Who Refused Meat", S["case_title"]),
hr(CASE_BORDER, 1.0),
Spacer(1, 0.1*cm),
Paragraph("<b>Patient:</b> Aisha, 3-year-old girl | <b>Setting:</b> Paediatric outpatient clinic", S["case_body"]),
Spacer(1, 0.15*cm),
Paragraph("<u>Presenting Complaint</u>", S["case_q"]),
Paragraph(
"Aisha is brought in by her parents with a 6-month history of <b>recurrent vomiting</b>, "
"<b>irritability</b>, and poor growth. Her parents note she consistently <b>refuses meat, "
"eggs, and legumes</b>, preferring only bread and fruits. Episodes of vomiting worsen "
"whenever she attends birthday parties where high-protein food is served.",
S["case_body"]
),
Spacer(1, 0.15*cm),
Paragraph("<u>Past History & Development</u>", S["case_q"]),
Paragraph(
"Born at term after an uncomplicated pregnancy. Normal neonatal screening (newborn markers "
"for HHH may be borderline or missed). Motor milestones slightly delayed: walked at "
"18 months. Vocabulary limited for age. One prior hospitalisation at age 2 for "
"'viral gastroenteritis with encephalopathy' — now thought to have been a hyperammonemic episode.",
S["case_body"]
),
Spacer(1, 0.15*cm),
Paragraph("<u>Examination Findings</u>", S["case_q"]),
Paragraph(
"Weight and height below the 3rd percentile. Mildly irritable. No jaundice. "
"Hepatomegaly (+2 cm below costal margin). Neurological exam: <b>brisk deep tendon "
"reflexes bilaterally</b>, mild clumsiness of gait. No focal deficit. "
"Fundoscopy normal.",
S["case_body"]
),
Spacer(1, 0.15*cm),
Paragraph("<u>Investigations</u>", S["case_q"]),
]
inv_rows = [
["Plasma ammonia", "128 µmol/L", "High (normal <50 µmol/L)"],
["Plasma ornithine", "410 µmol/L", "High (normal 40-120 µmol/L)"],
["Urine homocitrulline", "Markedly raised","Pathognomonic of HHH"],
["Plasma citrulline", "10 µmol/L", "Low (normal 15-40 µmol/L)"],
["Plasma glutamine", "Elevated", "Non-specific; reflects NH3 burden"],
["Urine orotic acid", "Normal", "Distinguishes from OTC deficiency"],
["Liver transaminases", "Mildly raised", "Common in urea cycle disorders"],
["MRI Brain", "Mild cerebral oedema", "Consistent with hyperammonemic injury"],
["SLC25A15 sequencing", "Homozygous c.535C>T (p.Arg179X)", "Confirms ORC1 deficiency"],
]
inv_t = Table(
[[Paragraph(r[0], S["tbl_cell"]),
Paragraph(f"<b>{r[1]}</b>", S["tbl_cell"]),
Paragraph(r[2], S["tbl_cell"])] for r in inv_rows],
colWidths=[4.5*cm, 4*cm, CONTENT_W - 8.5*cm - 1.6*cm]
)
inv_t.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,0), TABLE_HDR),
("TEXTCOLOR", (0,0), (-1,0), WHITE),
("ROWBACKGROUNDS",(0,1),(-1,-1), [WHITE, TABLE_ALT]),
("GRID", (0,0), (-1,-1), 0.4, GRAY_LINE),
("VALIGN", (0,0), (-1,-1), "TOP"),
("TOPPADDING", (0,0), (-1,-1), 5),
("BOTTOMPADDING",(0,0), (-1,-1), 5),
("LEFTPADDING", (0,0), (-1,-1), 7),
("RIGHTPADDING", (0,0), (-1,-1), 7),
]))
case_content.append(inv_t)
case_content.append(Spacer(1, 0.15*cm))
case_content += [
Paragraph("<u>Diagnosis</u>", S["case_q"]),
Paragraph(
"<b>HHH Syndrome (ORC1 deficiency)</b> — confirmed by the triad of hyperornithinemia, "
"hyperammonemia, homocitrullinuria, and SLC25A15 mutation.",
S["case_body"]
),
Spacer(1, 0.15*cm),
]
# Discussion Q&A
qas = [
("Q1. What is the single molecular defect responsible for all three abnormalities?",
"A. Loss-of-function mutation in SLC25A15 (ORC1), the mitochondrial ornithine/citrulline "
"antiporter. Without ORC1, ornithine cannot enter mitochondria (→ hyperornithinemia), the "
"urea cycle stalls (→ hyperammonemia), and carbamoyl phosphate reacts with lysine instead "
"(→ homocitrullinuria)."),
("Q2. Why is urine orotic acid NORMAL in HHH syndrome, unlike OTC deficiency?",
"A. In OTC deficiency, carbamoyl phosphate leaks into the cytosol and feeds pyrimidine "
"synthesis, producing excess orotic acid. In HHH syndrome, carbamoyl phosphate remains in "
"the mitochondria (where it reacts with lysine), so the cytosolic pyrimidine pathway is "
"not flooded — orotic acid remains normal."),
("Q3. Why does this child self-restrict protein intake?",
"A. Protein catabolism generates ammonia. In HHH syndrome, ammonia cannot be detoxified via "
"the urea cycle. The child's avoidance of high-protein foods is an instinctive protective "
"response to prevent hyperammonemic episodes — a classic behavioural clue in urea cycle disorders."),
("Q4. How should an acute hyperammonemic crisis be managed?",
"A. (1) Stop protein; (2) IV glucose + lipids ± insulin to halt catabolism; "
"(3) IV nitrogen scavengers (sodium benzoate + phenylacetate); "
"(4) IV arginine; (5) Haemodialysis if ammonia does not fall rapidly."),
("Q5. What is the specific long-term intervention unique to HHH syndrome?",
"A. Ornithine supplementation. By providing exogenous ornithine, some molecules diffuse "
"passively into mitochondria, partially restoring urea cycle flux — directly compensating "
"for the transport defect. This is in addition to the standard low-protein diet and "
"ammonia scavengers used in all urea cycle disorders."),
]
for q, a in qas:
case_content.append(Paragraph(q, S["case_q"]))
case_content.append(Paragraph(a, S["case_a"]))
case_content.append(Spacer(1, 0.05*cm))
case_content += [
Spacer(1, 0.1*cm),
Paragraph("<u>Management Plan for Aisha</u>", S["case_q"]),
Paragraph(
"<b>Acute phase:</b> IV glucose 10% + intralipid; sodium benzoate + sodium phenylacetate "
"(IV); arginine 200 mg/kg/day. Monitor ammonia every 2–4 hours. "
"<b>Discharge plan:</b> Protein-restricted diet (1.0–1.2 g/kg/day) as frequent "
"small meals; oral ornithine supplementation; oral sodium phenylbutyrate; "
"citrulline supplementation; regular neurological and developmental review. "
"<b>Long-term:</b> Refer to metabolic liver transplant programme for consideration "
"of liver transplantation.",
S["case_body"]
),
]
story.append(case_box(case_content))
story.append(Spacer(1, 0.4*cm))
# ── SECTION 9: INHERITANCE ────────────────────────────────────────────────────
story.append(section_header("9. Genetics & Inheritance"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph(
"HHH syndrome follows an <b>autosomal recessive</b> inheritance pattern. "
"Both copies of <i>SLC25A15</i> must carry loss-of-function mutations. "
"The gene encodes ORC1, a mitochondrial inner membrane solute carrier of the SLC25 family. "
"Considerable <b>phenotypic variability</b> exists even between patients sharing the same mutation, "
"suggesting modifier genes or environmental factors influence severity.",
S["body"]
))
gen_rows = [
["Inheritance", "Autosomal recessive"],
["Gene", "SLC25A15 (formerly ORC1)"],
["Protein", "Mitochondrial ornithine/citrulline antiporter (ORC1)"],
["Chromosomal locus", "13q14"],
["Carrier frequency", "Rare; higher in some isolated populations"],
["Phenotypic range", "Severe neonatal hyperammonemia to mild late-onset spastic paraparesis"],
]
story.append(two_col_table(gen_rows, ["Parameter", "Details"], [4*cm, CONTENT_W - 4*cm]))
# ── REFERENCES ─────────────────────────────────────────────────────────────────
story.append(Spacer(1, 0.5*cm))
story.append(hr(MED_BLUE, 1.5))
story.append(Paragraph("<b>References</b>", S["h2"]))
refs = [
"Harper's Illustrated Biochemistry, 32nd Ed — Ornithine Permease section, Chapter 28",
"Tietz Textbook of Laboratory Medicine, 7th Ed — Urea Cycle Defects, Chapter 60",
"Bradley and Daroff's Neurology in Clinical Practice — Hyperammonemia, Chapter 91",
"Adams and Victor's Principles of Neurology, 12th Ed — Inherited Hyperammonemias, Chapter 11",
"Emery's Elements of Medical Genetics and Genomics — Urea Cycle Disorders",
"OMIM #238970 — Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome",
]
for r in refs:
story.append(Paragraph(f"• {r}", S["ref"]))
# ── BUILD ─────────────────────────────────────────────────────────────────────
def add_page_number(canvas, doc):
canvas.saveState()
canvas.setFont("Helvetica", 8)
canvas.setFillColor(colors.grey)
canvas.drawRightString(
W - 2*cm, 1.5*cm,
f"HHH Syndrome — Medical Education | Page {doc.page}"
)
canvas.restoreState()
doc.build(story, onFirstPage=add_page_number, onLaterPages=add_page_number)
print(f"PDF saved to: {OUTPUT}")
cd /home/daytona/workspace/hhh-syndrome && python generate_pdf.py 2>&1
~/hhh-syndrome/generate_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/hhh-syndrome/generate_pdf.py'
cd /home/daytona/workspace/hhh-syndrome && python generate_pdf.py 2>&1
~/hhh-syndrome/generate_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/hhh-syndrome/generate_pdf.py'
~/hhh-syndrome/generate_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/hhh-syndrome/generate_pdf.py'
cd /home/daytona/workspace/hhh-syndrome && python generate_pdf.py 2>&1
ls -lh /home/daytona/workspace/hhh-syndrome/HHH_Syndrome.pdf
HHH Syndrome
PDF Document · PDF
- Normal Urea Cycle — contextual overview with the ORC1 transporter diagram (embedded from the textbook)
- The Primary Defect — ORC1/SLC25A15 mutation and what it disrupts
- Biochemical Mechanism — step-by-step table (4 steps): ornithine accumulation → urea cycle block → homocitrulline formation → brain toxicity
- Three Hallmarks Summary — colour-coded table linking each feature to its mechanism and lab marker
- Clinical Features — neonatal vs late-onset presentations
- Laboratory Diagnosis — full investigation table with expected values
- Treatment Plan — acute crisis management (5 steps) + long-term chronic management table
- 3-year-old girl (Aisha) with recurrent vomiting, protein aversion, developmental delay
- Full investigation results with values
- 5 discussion questions with model answers (ideal for classroom use)
- Complete management plan
Make a ppt for presentation in class on topic hyperammonemia 1 disorder . Explain proper mechanism about its biochemistry and treatment plan
carbamoyl phosphate synthetase I deficiency hyperammonemia type 1 urea cycle
mkdir -p /home/daytona/workspace/hyperammonemia-ppt && ls /home/daytona/workspac…
~/hyperammonemia-ppt/create_ppt.js
const pptxgen = require('pptxgenjs');
const pptx = new pptxgen();
pptx.layout = 'LAYOUT_WIDE'; // 13.3 x 7.5
pptx.author = 'Orris';
pptx.company = 'Medical Education';
pptx.subject = 'Hyperammonemia Type 1 (CPS1 Deficiency)';
pptx.title = 'Hyperammonemia Type 1 Disorder';
pptx.lang = 'en-US';
pptx.theme = {
headFontFace: 'Calibri',
bodyFontFace: 'Calibri',
lang: 'en-US'
};
const C = {
bg: '0F172A',
panel: '111827',
title: 'E2E8F0',
text: 'CBD5E1',
accent: '22D3EE',
accent2: '38BDF8',
warning: 'F59E0B',
danger: 'F43F5E',
good: '10B981',
white: 'FFFFFF',
black: '000000'
};
function addHeader(slide, title, subtitle='') {
slide.background = { color: C.bg };
slide.addShape(pptx.ShapeType.rect, {
x: 0, y: 0, w: 13.3, h: 0.9,
fill: { color: '0B1220' },
line: { color: '0B1220' }
});
slide.addText(title, {
x: 0.4, y: 0.18, w: 9.8, h: 0.4,
fontSize: 20, bold: true, color: C.title
});
if (subtitle) {
slide.addText(subtitle, {
x: 0.4, y: 0.55, w: 10.5, h: 0.25,
fontSize: 11, color: C.accent
});
}
slide.addShape(pptx.ShapeType.line, {
x: 0, y: 0.9, w: 13.3, h: 0,
line: { color: C.accent2, pt: 1 }
});
}
function addFooter(slide, text='Medical Biochemistry Class Presentation') {
slide.addShape(pptx.ShapeType.line, {
x: 0.35, y: 7.05, w: 12.6, h: 0,
line: { color: '334155', pt: 1 }
});
slide.addText(text, {
x: 0.4, y: 7.1, w: 12.4, h: 0.2,
fontSize: 9, color: '94A3B8', align: 'right'
});
}
function bullets(items) {
return items.map((t, i) => ({
text: t,
options: { bullet: true, breakLine: i !== items.length - 1 }
}));
}
// Slide 1: Title
{
const s = pptx.addSlide();
s.background = { color: C.bg };
s.addShape(pptx.ShapeType.rect, {
x: 0, y: 0, w: 13.3, h: 7.5,
fill: { color: '0B1220' }, line: { color: '0B1220' }
});
s.addShape(pptx.ShapeType.roundRect, {
x: 0.9, y: 0.95, w: 11.5, h: 5.2,
fill: { color: '111827', transparency: 8 },
line: { color: '1F2937', pt: 1.2, transparency: 0 },
radius: 0.08
});
s.addText('HYPERAMMONEMIA TYPE 1 DISORDER', {
x: 1.2, y: 1.6, w: 10.9, h: 0.9,
fontSize: 42, bold: true, color: C.title, align: 'center'
});
s.addText('Carbamoyl Phosphate Synthetase I (CPS1) Deficiency', {
x: 1.2, y: 2.65, w: 10.9, h: 0.5,
fontSize: 23, color: C.accent, bold: true, align: 'center'
});
s.addText('Biochemistry mechanism, diagnosis, and treatment plan', {
x: 1.2, y: 3.25, w: 10.9, h: 0.4,
fontSize: 16, color: C.text, align: 'center'
});
s.addText('Autosomal recessive urea cycle disorder | Hyperammonemia type 1', {
x: 1.2, y: 3.78, w: 10.9, h: 0.35,
fontSize: 13, color: '94A3B8', align: 'center'
});
s.addText('Prepared for class presentation', {
x: 1.2, y: 4.8, w: 10.9, h: 0.3,
fontSize: 13, color: '93C5FD', align: 'center', italic: true
});
addFooter(s);
}
// Slide 2: Learning objectives
{
const s = pptx.addSlide();
addHeader(s, 'Learning Objectives');
s.addText(bullets([
'Define Hyperammonemia Type 1 and identify the enzyme defect (CPS1).',
'Explain normal urea cycle nitrogen disposal and the role of N-acetylglutamate (NAG).',
'Describe the biochemical mechanism leading to hyperammonemia, glutamine accumulation, and encephalopathy.',
'Interpret key laboratory findings used for diagnosis and differentiation from other proximal urea cycle defects.',
'Present acute and long-term evidence-based treatment strategies for CPS1 deficiency.'
]), {
x: 0.9, y: 1.4, w: 11.7, h: 4.8,
fontSize: 20, color: C.text, valign: 'top', margin: 0.08,
breakLine: true
});
addFooter(s);
}
// Slide 3: Urea cycle overview
{
const s = pptx.addSlide();
addHeader(s, 'Normal Urea Cycle: Where CPS1 Fits');
s.addShape(pptx.ShapeType.roundRect, {
x: 0.6, y: 1.2, w: 5.95, h: 5.4,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }, radius: 0.06
});
s.addShape(pptx.ShapeType.roundRect, {
x: 6.75, y: 1.2, w: 5.95, h: 5.4,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }, radius: 0.06
});
s.addText('Mitochondrial steps', {
x: 0.95, y: 1.45, w: 5.2, h: 0.3,
fontSize: 16, bold: true, color: C.accent
});
s.addText(bullets([
'Step 1: CPS1 combines NH3 + HCO3− + 2 ATP → carbamoyl phosphate.',
'CPS1 is rate-limiting for entry of waste nitrogen into urea synthesis.',
'N-acetylglutamate (NAG) is an essential allosteric activator of CPS1.',
'Step 2: OTC uses carbamoyl phosphate + ornithine → citrulline.'
]), {
x: 0.95, y: 1.9, w: 5.3, h: 2.8,
fontSize: 14, color: C.text, valign: 'top', margin: 0.06
});
s.addText('Cytosolic steps', {
x: 7.1, y: 1.45, w: 5.2, h: 0.3,
fontSize: 16, bold: true, color: C.accent
});
s.addText(bullets([
'Citrulline + aspartate → argininosuccinate (ASS).',
'Argininosuccinate → arginine + fumarate (ASL).',
'Arginine → urea + ornithine (arginase).',
'One nitrogen of urea comes from free ammonia, one from aspartate.'
]), {
x: 7.1, y: 1.9, w: 5.3, h: 2.8,
fontSize: 14, color: C.text, valign: 'top', margin: 0.06
});
s.addShape(pptx.ShapeType.roundRect, {
x: 0.95, y: 4.95, w: 11.3, h: 1.3,
fill: { color: '082F49' }, line: { color: '0EA5E9', pt: 1 }, radius: 0.05
});
s.addText('Key biochemical point: NAG is essential for CPS1 activity.\nWithout CPS1, ammonia cannot be incorporated into carbamoyl phosphate, so the urea cycle cannot start.', {
x: 1.15, y: 5.18, w: 10.9, h: 0.9,
fontSize: 14, bold: true, color: 'E0F2FE', align: 'center', valign: 'mid'
});
addFooter(s);
}
// Slide 4: What is Hyperammonemia type 1
{
const s = pptx.addSlide();
addHeader(s, 'Hyperammonemia Type 1: Definition and Genetics');
s.addText('Definition', {
x: 0.8, y: 1.25, w: 3.5, h: 0.4,
fontSize: 18, bold: true, color: C.accent
});
s.addText('Hyperammonemia Type 1 is a proximal urea cycle disorder caused by deficiency of mitochondrial carbamoyl phosphate synthetase I (CPS1).', {
x: 0.8, y: 1.65, w: 12.0, h: 0.9,
fontSize: 17, color: C.text
});
s.addTable([
[
{ text: 'Feature', options: { bold: true, color: C.white, fill: { color: '0369A1' } } },
{ text: 'Hyperammonemia Type 1 (CPS1 deficiency)', options: { bold: true, color: C.white, fill: { color: '0369A1' } } }
],
['Defective enzyme', 'Carbamoyl phosphate synthetase I (CPS1)'],
['Inheritance', 'Autosomal recessive'],
['Pathway location', 'Mitochondrial first committed step of urea cycle'],
['Biochemical hallmark', 'Severe hyperammonemia with low citrulline and low/normal orotic acid'],
['Typical onset', 'Neonatal severe form or later-onset partial deficiency']
], {
x: 0.8, y: 2.65, w: 12.0, h: 3.2,
colW: [3.3, 8.7],
border: { pt: 1, color: '334155' },
fontSize: 13,
color: C.text,
fill: '111827',
valign: 'mid',
margin: 0.05
});
addFooter(s);
}
// Slide 5: Biochemical pathogenesis flow
{
const s = pptx.addSlide();
addHeader(s, 'Biochemical Mechanism of Disease');
const steps = [
{ t: '1. CPS1 activity is absent or markedly reduced', c: 'Loss of CPS1 prevents NH3 fixation into carbamoyl phosphate.' },
{ t: '2. Urea cycle entry is blocked at reaction 1', c: 'Downstream intermediates (especially citrulline/arginine) become deficient.' },
{ t: '3. Ammonia accumulates rapidly in blood', c: 'Nitrogen disposal fails, especially during protein load or catabolic stress.' },
{ t: '4. Brain converts NH3 + glutamate → glutamine', c: 'Glutamine accumulates in astrocytes causing osmotic swelling and cerebral edema.' },
{ t: '5. Clinical encephalopathy develops', c: 'Irritability, vomiting, lethargy, seizures, coma, and possible death if untreated.' }
];
let y = 1.3;
steps.forEach((st, idx) => {
s.addShape(pptx.ShapeType.roundRect, {
x: 0.8, y, w: 12.0, h: 1.0,
fill: { color: idx % 2 === 0 ? '0B3B5A' : '12324A' },
line: { color: '334155', pt: 0.8 },
radius: 0.04
});
s.addText(st.t, {
x: 1.0, y: y + 0.12, w: 11.6, h: 0.28,
fontSize: 15, bold: true, color: 'BAE6FD'
});
s.addText(st.c, {
x: 1.0, y: y + 0.42, w: 11.6, h: 0.42,
fontSize: 12.5, color: C.text
});
y += 1.13;
});
addFooter(s);
}
// Slide 6: Why neurological damage occurs
{
const s = pptx.addSlide();
addHeader(s, 'Neurotoxicity in Hyperammonemia Type 1');
s.addShape(pptx.ShapeType.roundRect, {
x: 0.8, y: 1.2, w: 6.0, h: 5.7,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }
});
s.addShape(pptx.ShapeType.roundRect, {
x: 6.95, y: 1.2, w: 5.55, h: 5.7,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }
});
s.addText('Core biochemical events', {
x: 1.1, y: 1.45, w: 5.4, h: 0.3,
fontSize: 16, bold: true, color: C.accent
});
s.addText(bullets([
'High NH3 crosses blood-brain barrier rapidly.',
'Astrocytes detoxify NH3 by glutamine synthetase.',
'Excess glutamine increases intracellular osmotic load.',
'Astrocyte swelling causes cerebral edema and raised intracranial pressure.',
'Glutamate handling and neuronal signaling are disturbed.',
'Severe episodes can cause permanent cognitive and motor deficits.'
]), {
x: 1.1, y: 1.9, w: 5.3, h: 4.7,
fontSize: 13.4, color: C.text, margin: 0.05
});
s.addText('Clinical correlates', {
x: 7.2, y: 1.45, w: 4.9, h: 0.3,
fontSize: 16, bold: true, color: C.warning
});
s.addText(bullets([
'Early: poor feeding, vomiting, tachypnea, irritability.',
'Progression: lethargy, hypotonia, seizures, coma.',
'Acid-base: respiratory alkalosis is common in acute episodes.',
'Long term: developmental delay, intellectual disability, behavioral issues.'
]), {
x: 7.2, y: 1.9, w: 5.0, h: 2.7,
fontSize: 13.2, color: C.text, margin: 0.05
});
s.addShape(pptx.ShapeType.roundRect, {
x: 7.2, y: 4.95, w: 5.0, h: 1.45,
fill: { color: '3F1D2E' }, line: { color: C.danger, pt: 1 }
});
s.addText('Emergency principle:\nEvery hour of uncontrolled ammonia increases risk of irreversible brain injury.', {
x: 7.4, y: 5.2, w: 4.65, h: 1.0,
fontSize: 13, bold: true, color: 'FECDD3', align: 'center', valign: 'mid'
});
addFooter(s);
}
// Slide 7: Clinical presentation
{
const s = pptx.addSlide();
addHeader(s, 'Clinical Presentation Across Age Groups');
s.addTable([
[
{ text: 'Age Group', options: { bold: true, color: C.white, fill: { color: '075985' } } },
{ text: 'Typical Presentation', options: { bold: true, color: C.white, fill: { color: '075985' } } },
{ text: 'Common Triggers', options: { bold: true, color: C.white, fill: { color: '075985' } } }
],
[
'Neonatal severe form',
'Initially well newborn, then poor feeding, vomiting, tachypnea, lethargy, seizures, coma in 24–72 h',
'Postnatal protein intake and physiologic catabolic transition'
],
[
'Infancy/childhood partial deficiency',
'Recurrent vomiting, episodic confusion, developmental delay, protein aversion',
'Infection, fasting, high-protein meals, surgery'
],
[
'Adolescents/adults (mild variants)',
'Intermittent encephalopathy, headache, psychiatric/behavioral changes, cyclic vomiting',
'Stress, postpartum period, medications, catabolic illness'
]
], {
x: 0.7, y: 1.35, w: 12.1, h: 3.9,
colW: [2.7, 6.0, 3.4],
border: { pt: 1, color: '334155' },
color: C.text, fontSize: 12.5,
fill: '111827', margin: 0.04, valign: 'mid'
});
s.addShape(pptx.ShapeType.roundRect, {
x: 0.8, y: 5.55, w: 12.0, h: 1.1,
fill: { color: '052E16' }, line: { color: C.good, pt: 1 }
});
s.addText('Clinical clue: unexplained encephalopathy with respiratory alkalosis and no sepsis should trigger urgent ammonia testing.', {
x: 1.0, y: 5.9, w: 11.6, h: 0.5,
fontSize: 14, bold: true, color: 'BBF7D0', align: 'center'
});
addFooter(s);
}
// Slide 8: Diagnostic strategy
{
const s = pptx.addSlide();
addHeader(s, 'Diagnostic Workup and Differential Diagnosis');
s.addText('Essential tests in suspected CPS1 deficiency', {
x: 0.75, y: 1.2, w: 6.5, h: 0.35,
fontSize: 16, bold: true, color: C.accent
});
s.addText(bullets([
'Plasma ammonia: markedly elevated (often >150–200 umol/L in severe episodes).',
'Plasma amino acids: low citrulline, low arginine, elevated glutamine.',
'Urine orotic acid: typically low/normal (helps separate from OTC deficiency).',
'Liver function profile and blood gas (respiratory alkalosis may be present).',
'Confirmatory molecular testing: CPS1 gene sequencing.'
]), {
x: 0.85, y: 1.65, w: 6.15, h: 4.2,
fontSize: 13.5, color: C.text, margin: 0.05
});
s.addShape(pptx.ShapeType.roundRect, {
x: 7.15, y: 1.2, w: 5.45, h: 5.6,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }
});
s.addText('Differentiate proximal UCDs', {
x: 7.45, y: 1.45, w: 4.9, h: 0.35,
fontSize: 15, bold: true, color: C.warning
});
s.addTable([
[
{ text: 'Condition', options: { bold: true, color: C.white, fill: { color: '7C2D12' } } },
{ text: 'Citrulline', options: { bold: true, color: C.white, fill: { color: '7C2D12' } } },
{ text: 'Orotic Acid', options: { bold: true, color: C.white, fill: { color: '7C2D12' } } }
],
['CPS1 deficiency', 'Low', 'Low/Normal'],
['NAGS deficiency', 'Low', 'Low/Normal'],
['OTC deficiency', 'Low', 'High']
], {
x: 7.45, y: 2.0, w: 4.9, h: 2.0,
colW: [2.1, 1.2, 1.6],
border: { pt: 1, color: '334155' },
fill: '0F172A', color: C.text, fontSize: 11.5, margin: 0.03
});
s.addText('Important caution: NAGS deficiency can look biochemically identical to CPS1 deficiency, but responds to N-carbamylglutamate.', {
x: 7.45, y: 4.3, w: 4.9, h: 1.8,
fontSize: 12, color: 'FDE68A', italic: true, valign: 'top'
});
addFooter(s);
}
// Slide 9: Acute treatment plan
{
const s = pptx.addSlide();
addHeader(s, 'Acute Management (Medical Emergency)');
const leftX = 0.7;
const boxW = 12.0;
const boxH = 0.86;
const startY = 1.2;
const acuteSteps = [
'1) Stop protein intake immediately (temporary).',
'2) Reverse catabolism: high-calorie IV dextrose ± lipids; insulin if needed.',
'3) Start nitrogen scavengers: sodium benzoate and sodium phenylacetate/phenylbutyrate.',
'4) Give arginine (unless contraindicated) to support downstream nitrogen handling and protein synthesis.',
'5) Monitor ammonia every 2–4 hours and neurologic status closely.',
'6) Initiate hemodialysis rapidly if ammonia remains high or neurologic deterioration occurs.'
];
acuteSteps.forEach((txt, i) => {
s.addShape(pptx.ShapeType.roundRect, {
x: leftX, y: startY + i*(boxH+0.13), w: boxW, h: boxH,
fill: { color: i % 2 === 0 ? '082F49' : '0C4A6E' },
line: { color: '334155', pt: 0.8 }, radius: 0.05
});
s.addText(txt, {
x: leftX + 0.25, y: startY + i*(boxH+0.13) + 0.22, w: boxW-0.45, h: 0.45,
fontSize: 14, bold: i===5, color: 'E0F2FE'
});
});
s.addShape(pptx.ShapeType.roundRect, {
x: 0.95, y: 6.78, w: 11.5, h: 0.55,
fill: { color: '3F1D2E' }, line: { color: C.danger, pt: 1 }
});
s.addText('Dialysis is the fastest way to lower toxic ammonia when medical therapy is insufficient.', {
x: 1.2, y: 6.94, w: 11.0, h: 0.22,
fontSize: 12.5, bold: true, color: 'FECDD3', align: 'center'
});
addFooter(s);
}
// Slide 10: Long-term management
{
const s = pptx.addSlide();
addHeader(s, 'Long-Term Treatment and Follow-up');
s.addTable([
[
{ text: 'Intervention', options: { bold: true, color: C.white, fill: { color: '0369A1' } } },
{ text: 'Biochemical Rationale', options: { bold: true, color: C.white, fill: { color: '0369A1' } } },
{ text: 'Practical Notes', options: { bold: true, color: C.white, fill: { color: '0369A1' } } }
],
[
'Protein-controlled diet',
'Reduces nitrogen influx and ammonia generation',
'Use adequate calories to avoid catabolism; dietician-guided plan'
],
[
'Essential amino acid / citrulline support',
'Maintains growth and supports nitrogen handling downstream',
'Dose individualized by metabolic team'
],
[
'Oral ammonia scavengers',
'Alternative nitrogen excretion via conjugation pathways',
'Phenylbutyrate / glycerol phenylbutyrate / benzoate'
],
[
'Emergency sick-day protocol',
'Prevents catabolic surges during infection/fasting',
'Early hospital contact and rapid calorie rescue'
],
[
'Liver transplantation',
'Provides functional hepatic urea cycle and prevents recurrent crises',
'Consider in severe/refractory cases'
],
[
'Neurodevelopmental follow-up',
'Detects consequences of prior hyperammonemia early',
'Cognitive, motor, and behavioral monitoring'
]
], {
x: 0.6, y: 1.25, w: 12.2, h: 5.8,
colW: [3.2, 4.8, 4.2],
border: { pt: 1, color: '334155' },
fill: '111827',
color: C.text,
fontSize: 12,
valign: 'mid',
margin: 0.04
});
addFooter(s);
}
// Slide 11: Quick case-based slide
{
const s = pptx.addSlide();
addHeader(s, 'Clinical Case (Class Discussion)');
s.addShape(pptx.ShapeType.roundRect, {
x: 0.7, y: 1.2, w: 12.0, h: 2.35,
fill: { color: '1F2937' }, line: { color: '334155', pt: 1 }, radius: 0.06
});
s.addText('Case vignette', {
x: 0.95, y: 1.45, w: 2.0, h: 0.3,
fontSize: 15, bold: true, color: C.accent
});
s.addText('A 3-day-old term newborn develops poor feeding, tachypnea, vomiting, and progressive lethargy. Sepsis screen is negative. Blood gas shows respiratory alkalosis. Plasma ammonia is 420 umol/L. Plasma citrulline is very low and urine orotic acid is low.', {
x: 0.95, y: 1.82, w: 11.5, h: 1.45,
fontSize: 13.5, color: C.text, valign: 'top'
});
s.addShape(pptx.ShapeType.roundRect, {
x: 0.7, y: 3.85, w: 5.85, h: 2.85,
fill: { color: '082F49' }, line: { color: '0EA5E9', pt: 1 }, radius: 0.06
});
s.addText('Interpretation', {
x: 0.95, y: 4.05, w: 2.7, h: 0.3,
fontSize: 14.5, bold: true, color: 'BAE6FD'
});
s.addText(bullets([
'Pattern strongly suggests proximal UCD.',
'Low orotic acid makes OTC deficiency less likely.',
'Most likely diagnosis: CPS1 deficiency (Hyperammonemia Type 1).'
]), {
x: 0.95, y: 4.4, w: 5.35, h: 2.1,
fontSize: 12.6, color: C.text, margin: 0.05
});
s.addShape(pptx.ShapeType.roundRect, {
x: 6.85, y: 3.85, w: 5.85, h: 2.85,
fill: { color: '3F1D2E' }, line: { color: C.danger, pt: 1 }, radius: 0.06
});
s.addText('Immediate actions', {
x: 7.1, y: 4.05, w: 2.8, h: 0.3,
fontSize: 14.5, bold: true, color: 'FECDD3'
});
s.addText(bullets([
'Stop protein, start high-calorie IV dextrose.',
'Begin ammonia scavengers + arginine urgently.',
'Prepare for hemodialysis if ammonia does not rapidly fall.'
]), {
x: 7.1, y: 4.4, w: 5.35, h: 2.1,
fontSize: 12.6, color: 'FEE2E2', margin: 0.05
});
addFooter(s);
}
// Slide 12: Summary + references
{
const s = pptx.addSlide();
addHeader(s, 'Take-Home Messages and References');
s.addText('Take-home messages', {
x: 0.8, y: 1.2, w: 5.4, h: 0.4,
fontSize: 17, bold: true, color: C.accent
});
s.addText(bullets([
'Hyperammonemia Type 1 is CPS1 deficiency: a proximal block at urea cycle entry.',
'NAG is obligatory for CPS1 activity; without CPS1, ammonia detoxification fails.',
'Neurotoxicity is driven by ammonia and glutamine-mediated cerebral edema.',
'Biochemical profile: high ammonia, low citrulline, low/normal orotic acid.',
'Treatment has two goals: rapid ammonia reduction and prevention of future catabolic crises.'
]), {
x: 0.9, y: 1.7, w: 6.0, h: 3.9,
fontSize: 13.3, color: C.text, margin: 0.05
});
s.addShape(pptx.ShapeType.roundRect, {
x: 7.0, y: 1.2, w: 5.5, h: 5.4,
fill: { color: '111827' }, line: { color: '334155', pt: 1 }, radius: 0.06
});
s.addText('References', {
x: 7.25, y: 1.45, w: 2.8, h: 0.35,
fontSize: 16, bold: true, color: C.warning
});
const refs = [
"Harper's Illustrated Biochemistry, 32nd Ed, Ch. 28.",
"Lippincott Illustrated Reviews: Biochemistry, 8th Ed, Ch. 19.",
"Tietz Textbook of Laboratory Medicine, 7th Ed, Urea Cycle Defects.",
"Harrison's Principles of Internal Medicine, 22E, Urea Cycle Defects.",
"Genetic and biochemical consensus guidance on UCD emergency management."
];
s.addText(refs.map((r, i) => ({ text: `${i+1}. ${r}`, options: { breakLine: i !== refs.length - 1 } })), {
x: 7.3, y: 1.95, w: 4.9, h: 4.3,
fontSize: 11.6, color: C.text, margin: 0.03, valign: 'top'
});
s.addText('Thank you', {
x: 0.85, y: 6.35, w: 6.0, h: 0.55,
fontSize: 28, bold: true, color: 'BFDBFE'
});
addFooter(s, 'Prepared for teaching use | Hyperammonemia Type 1');
}
pptx.writeFile({ fileName: '/home/daytona/workspace/hyperammonemia-ppt/Hyperammonemia_Type1_Presentation.pptx' });
node /home/daytona/workspace/hyperammonemia-ppt/create_ppt.js
ls -lh /home/daytona/workspace/hyperammonemia-ppt/Hyperammonemia_Type1_Presentat…
Hyperammonemia Type1 Presentation
Presentation · PPTX
- clear urea cycle biochemistry
- CPS1/NAG mechanism
- stepwise disease pathogenesis
- neurotoxicity explanation
- diagnostic differentiation (CPS1 vs OTC vs NAGS)
- acute emergency treatment protocol
- long-term treatment and follow-up plan
- one case-discussion slide
- summary + references slide