CLL explains pathophisiology
Reading File
Finding Sources
Searching PubMed
"chronic lymphocytic leukemia" AND pathophysiology
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Loading Image
I now have everything needed to compose a comprehensive answer. Here is the full pathophysiology of CLL based on Harrison's Principles of Internal Medicine 22E and Goldman-Cecil Medicine.
Chronic Lymphocytic Leukemia (CLL) — Pathophysiology
Definition
CLL is a monoclonal proliferation of mature B lymphocytes defined by an absolute count of malignant cells in the blood ≥5 × 10⁹/L. Below this threshold, when there is no nodal, splenic, or hepatic involvement and no cytopenias, the condition is classified as monoclonal B-cell lymphocytosis (MBL), which carries a ~1–2% annual risk of progressing to overt CLL. — Harrison's Principles of Internal Medicine 22E, p. 893
The peripheral blood smear shows the characteristic small, mature-appearing lymphocytes:
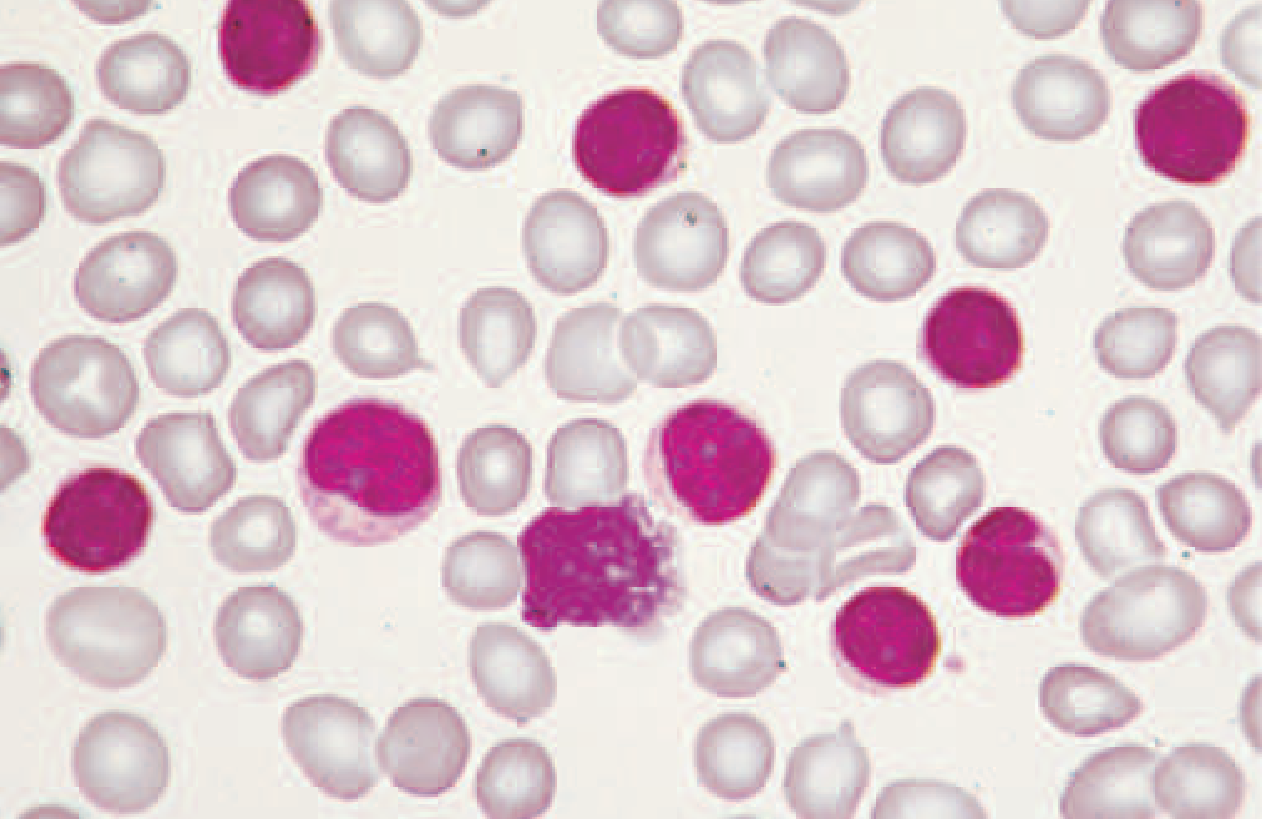
1. Cell of Origin
The cell of origin has not been definitively established. The morphology, immunophenotype, and gene expression pattern of CLL cells resemble a mature B cell, and memory B cells have historically been presumed as the progenitor. However, several features challenge this:
- The antigen-binding characteristics of CLL B-cell receptors (BCRs) and the presence of stereotyped BCRs (identical or near-identical variable-region sequences across unrelated patients) suggest a role for antigen selection, not simple mature lymphocyte expansion.
- Transplant experiments in mice where hematopoietic stem cells (HSCs) from CLL patients produced leukemia with characteristics differing from the donor CLL suggest HSC involvement.
- The most likely scenario is a stepwise transforming process at multiple stages of B-cell development, potentially including de-differentiation. — Harrison's 22E, p. 893
2. B-Cell Receptor (BCR) Signaling — The Central Driver
BCR signaling is the most important mechanistic advance in understanding CLL:
- CLL cells have distinct BCR signaling compared to normal B cells: low-level IgM surface expression, variable response to antigen stimulation, and tonic (constitutive) activation of antiapoptotic signaling pathways that promote tumor cell survival.
- Gene expression profiling shows CLL cells share features with antigen-activated mature B cells, implicating BCR activation in pathogenesis.
- In lymph nodes and bone marrow (vs. peripheral blood), BCR pathway genes are upregulated, making the microenvironment a particularly active site of tumor promotion.
- The crucial downstream effector is Bruton's tyrosine kinase (BTK), which propagates BCR signals to activate NF-κB, PI3K, and ERK pathways — explaining why BTK inhibitors (ibrutinib, acalabrutinib, zanubrutinib) are so effective. — Harrison's 22E, p. 893–894
3. IGHV Mutational Status — Two Disease Subtypes
During normal B-cell maturation, the variable regions of the immunoglobulin heavy chain undergo somatic hypermutation in germinal centers. This creates two biologically distinct CLL subtypes:
| Feature | IGHV mutated (~60%) | IGHV unmutated (~40%) |
|---|---|---|
| Germline deviation | ≥2% mutated | <2% from germline |
| Suggested progenitor | Post–germinal center (memory B cell) | Pre–germinal center |
| Clinical course | Indolent, longer survival | Aggressive, rapid progression |
| BCR signaling | Lower tonic activity | Higher, autonomous tonic activation |
| Associated mutations | Fewer high-risk mutations | NOTCH1, SF3B1, TP53, ATM more common |
| Response to chemotherapy | Better (FCR can achieve long-term remission) | Inferior |
Additionally, ~30% of CLL patients express "stereotyped" BCRs — nearly identical receptor sequences across unrelated patients — implying that common antigens (possibly self-antigens or microbial) drive clonal selection. — Harrison's 22E, p. 893
4. Cytogenetic Abnormalities
Detected by FISH analysis, these are the most robust prognostic markers:
| Abnormality | Frequency | Gene Affected | Prognosis |
|---|---|---|---|
| del(13)(q14.3) — sole | ~55% | miR-15a/16-1 | Favorable — most indolent |
| Trisomy 12 | ~15% | — | Intermediate |
| del(11)(q22.3) | ~10–15% | ATM | Poor — bulky disease, aggressive |
| del(17)(p13.1) | ~5–10% | TP53 | Worst — chemo-resistant, rapid progression |
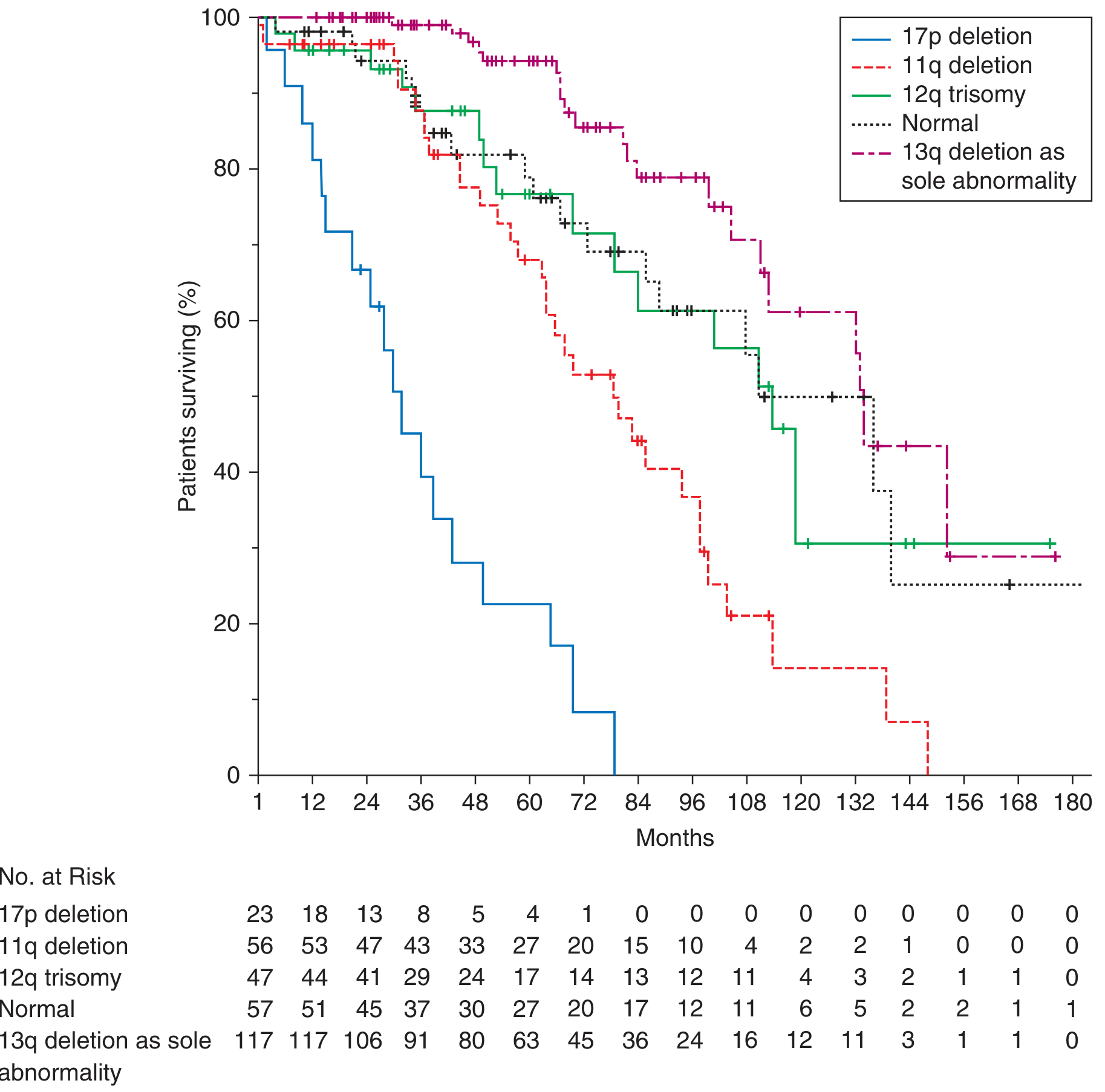
- del(13q14.3): Deletes the miR-15a/miR-16-1 microRNA cluster, which normally suppresses BCL2. Their loss → BCL2 overexpression → impaired apoptosis.
- del(17p) / TP53 loss: Disables the DNA damage checkpoint. Cells cannot undergo apoptosis after genotoxic stress → resistance to chemotherapy. When combined with a TP53 point mutation on the other allele, both copies are inactivated.
- del(11q) / ATM loss: ATM normally activates TP53 in response to DNA double-strand breaks. Its deletion → impaired DNA damage response.
- Clonal evolution: Acquisition of new cytogenetic abnormalities is common, especially in IGHV-unmutated disease, and FISH should be repeated before every line of therapy. — Harrison's 22E, p. 894
5. Recurrent Gene Mutations
Unlike many hematologic malignancies, CLL has no unifying driver mutation; the average CLL genome carries ~20 nonsynonymous alterations. Key mutations:
| Gene | Frequency | Mechanism |
|---|---|---|
| SF3B1 | ~10–15% | RNA spliceosome component; aberrant splicing of tumor suppressors |
| NOTCH1 | ~10–15% | PEST domain mutations → constitutive NOTCH signaling → survival, anti-apoptosis; associated with trisomy 12 and Richter transformation |
| MYD88 | ~3–5% | Toll-like receptor adaptor → NF-κB activation |
| ATM | ~10% | DNA damage response (also deleted in del 11q) |
| TP53 | ~5–8% (higher in relapsed) | Tumor suppressor (also deleted in del 17p) |
NOTCH1 mutations are notably linked to Richter's transformation — the conversion of CLL to aggressive diffuse large B-cell lymphoma (DLBCL), which occurs in ~2–10% of patients. — Harrison's 22E, p. 894
6. Antiapoptotic Mechanisms — BCL2 Overexpression
A hallmark of CLL is failure of programmed cell death:
- Loss of miR-15a/16-1 (del 13q) removes post-transcriptional suppression of BCL2.
- BCL2 protein sequesters pro-apoptotic factors (BAX, BAK), preventing cytochrome c release and caspase activation.
- This makes CLL cells long-lived despite being relatively non-proliferative — the clone expands primarily by impaired cell death, not by rapid proliferation.
- This mechanism is therapeutically exploited by venetoclax, a selective BCL2 inhibitor. — Harrison's 22E, p. 895
7. Tumor Microenvironment
CLL cells do not survive in isolation — the bone marrow and lymph node microenvironments actively support tumor cell survival:
- Nurse-like cells (NLCs) and stromal cells in the bone marrow provide survival signals via CXCL12/CXCL13 chemokines.
- T-regulatory cells (Tregs) and myeloid-derived suppressor cells impair antitumor immune responses.
- CD4+ T cells in CLL form abnormal immunological synapses with CLL B cells, promoting tumor survival rather than tumor killing.
- CD38 and ZAP-70 on the CLL cell surface are markers of active BCR signaling and interaction with the microenvironment; their expression correlates with IGHV unmutated status and poor prognosis. — Harrison's 22E; Goldman-Cecil Medicine, p. 1928
8. Immune Dysfunction
CLL causes profound immunosuppression that is both intrinsic to the disease and compounded by therapy:
- Hypogammaglobulinemia: Reduced normal B-cell function → decreased Ig production → susceptibility to encapsulated bacteria (pneumonia, sinusitis).
- T-cell dysfunction: Inverted CD4:CD8 ratio, exhausted T-cell phenotype (upregulation of PD-1, CTLA-4).
- NK cell impairment: Reduced cytotoxic killing of tumor cells.
- Infections account for ~30–50% of all CLL-related deaths. — Harrison's 22E, p. 896
Immunophenotype (distinguishing CLL from other B-cell malignancies):
| Marker | CLL | Mantle Cell | Marginal Zone | Follicular |
|---|---|---|---|---|
| CD5 | + | + | −/+ | − |
| CD10 | − | − | − | + |
| CD19 | + | + | + | + |
| CD20 | dim | mod/bright | mod/bright | + |
| CD23 | + | − | −/+ | + |
| Cyclin D1 | − | + | − | − |
| Surface Ig | dim | mod/bright | mod/bright | — |
The CD5+/CD23+/dim CD20/dim surface Ig combination is the classic CLL immunophenotype. — Harrison's 22E, p. 896
Summary: Integrated Pathophysiologic Model
Genetic predisposition (GWAS SNPs, familial risk)
↓
Initiating event in B-cell progenitor (HSC or mature B cell)
↓
Clonal expansion driven by:
• Tonic BCR signaling (BTK → NF-κB, PI3K)
• Antigen selection (stereotyped BCRs)
• Cytogenetic lesions (del 13q → BCL2↑; del 17p → TP53 loss)
• Gene mutations (NOTCH1, SF3B1, ATM, MYD88)
↓
Impaired apoptosis (BCL2 overexpression)
↓
Microenvironment support (NLCs, Tregs, CXCLs in BM/LN)
↓
Tumor accumulation in blood, bone marrow, lymph nodes, spleen
↓
Clinical consequences:
• Lymphocytosis, lymphadenopathy, splenomegaly
• Bone marrow failure → cytopenias
• Immune dysfunction → infections, autoimmune cytopenias
• Richter's transformation (~2–10%)
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 112 — Chronic Lymphocytic Leukemia
- Goldman-Cecil Medicine, Chapter on CLL — Pathobiology, p. 1928
Treatment
Reading File
Searching PubMed
"chronic lymphocytic leukemia" AND treatment
Reading File
Reading Article
Reading File
Reading File
I now have everything needed for a comprehensive treatment overview. Here is the full CLL treatment guide.
CLL — Treatment
1. When to Treat: "Watch and Wait"
CLL does not require immediate treatment at diagnosis. Multiple trials have shown no survival advantage with early therapy. Treatment is initiated only when symptomatic disease develops (iwCLL criteria):
| Indication for Treatment |
|---|
| Evidence of progressive marrow failure (worsening anemia or thrombocytopenia from BM involvement) |
| Massive splenomegaly (≥6 cm below costal margin), progressive, or symptomatic |
| Massive lymphadenopathy (≥10 cm), progressive, or symptomatic |
| Progressive lymphocytosis: ≥50% increase over 2 months OR lymphocyte doubling time <6 months |
| Autoimmune cytopenia not responsive to standard therapy |
| Symptomatic or functional extramedullary disease |
| Significant disease-related constitutional symptoms (fatigue, night sweats, fever, weight loss) |
Harrison's 22E, p. 898–899
2. Staging Before Treatment
Rai Staging (US)
| Stage | Description |
|---|---|
| 0 (Low risk) | Lymphocytosis only |
| I/II (Intermediate) | Lymphocytosis + lymphadenopathy ± splenomegaly/hepatomegaly |
| III/IV (High risk) | Lymphocytosis + anemia or thrombocytopenia (BM involvement) |
Binet Staging (Europe)
| Stage | Description |
|---|---|
| A | <3 areas of lymphadenopathy |
| B | ≥3 areas of lymphadenopathy |
| C | Hgb ≤10 g/dL and/or platelets <100,000/μL |
CLL International Prognostic Index (CLL-IPI)
| Variable | Adverse Factor | Score |
|---|---|---|
| TP53 status | Deleted or mutated | 4 |
| IGHV status | Unmutated | 2 |
| β2-microglobulin | >3.5 mg/L | 2 |
| Clinical stage | Rai I–IV or Binet B–C | 1 |
| Age | >65 years | 1 |
| Total Score | Risk | 5-Year Survival |
|---|---|---|
| 0–1 | Low | 93.2% |
| 2–3 | Intermediate | 79.3% |
| 4–6 | High | 63.3% |
| 7–10 | Very high | 23.3% |
Harrison's 22E, p. 898
3. First-Line Treatment — Targeted Therapies (Current Standard)
Over the past decade, oral targeted therapies have replaced chemoimmunotherapy as the standard of care for most patients. Treatment is built around two key vulnerabilities in CLL biology: BTK (the BCR signaling kinase) and BCL2 (the antiapoptotic protein).
A. BTK Inhibitors
BTK is an ideal target because it has no natural redundancy and is relatively B-cell selective.
| Drug | Generation | Key Trials | Key Data |
|---|---|---|---|
| Ibrutinib | 1st (covalent) | RESONATE, E1912 | 84% lower risk of progression vs. chlorambucil; 59% PFS at 7 years |
| Acalabrutinib | 2nd (covalent) | ELEVATE-TN, ELEVATE-RR | 6-year PFS 62% alone; 78% PFS at 4 years + obinutuzumab |
| Zanubrutinib | 2nd (covalent) | SEQUOIA, ALPINE | 85.5% PFS at 2 years frontline; superior PFS vs. ibrutinib in ALPINE |
Mechanism: Irreversibly block BTK → inhibit BCR-mediated NF-κB, PI3K, ERK signaling → impair CLL cell survival, proliferation, and microenvironmental homing.
Second-generation agents (acalabrutinib, zanubrutinib) are now preferred over ibrutinib due to greater BTK selectivity and better tolerability (lower rates of atrial fibrillation, hypertension, arthralgias).
Side effects of BTK inhibitors:
- Atrial fibrillation / ventricular arrhythmias (ibrutinib > second-generation)
- Hypertension
- Bleeding / bruising (antiplatelet effect via TEC kinase inhibition)
- Arthralgias, myalgias, rash, diarrhea
- Infections
Important prognostic nuance: With BTK inhibitors, traditional risk factors (IGHV status, FISH) have less impact on outcome — in the ELEVATE-TN trial, only age and performance status predicted survival.
B. BCL2 Inhibitor — Venetoclax
Mechanism: Orally bioavailable, selective allosteric BCL2 inhibitor → frees pro-apoptotic BAX/BAK → releases cytochrome c → caspase cascade → apoptosis of CLL cells.
Key regimens:
- VO (venetoclax + obinutuzumab): Fixed 1-year duration. CLL14 trial: median PFS 76.2 months vs. 36.4 months for chlorambucil + obinutuzumab. Approved frontline for unfit patients.
- VR (venetoclax + rituximab): For relapsed/refractory CLL; fixed 2-year duration.
- IVO (ibrutinib + venetoclax + obinutuzumab): CLL13/GAIA: superior PFS vs. chemoimmunotherapy (85.5% PFS at 4 years).
Side effects:
- Tumor lysis syndrome (TLS) — key concern; requires ramp-up dosing and monitoring
- Neutropenia
- Nausea/vomiting/diarrhea
C. Anti-CD20 Monoclonal Antibodies
Used in combination (not as monotherapy in CLL):
| Drug | Type | Notes |
|---|---|---|
| Rituximab | Anti-CD20, chimeric | Used with FCR, BR, or venetoclax |
| Obinutuzumab | Anti-CD20, humanized, glycoengineered | Preferred over rituximab with venetoclax (superior efficacy) |
D. Chemoimmunotherapy (Now Reserved for Selected Patients)
FCR (fludarabine + cyclophosphamide + rituximab): Once the gold standard, now mainly for young, fit patients with IGHV-mutated disease — ~15% can achieve durable remissions >20 years (possible cure in this subgroup). E1912 trial showed no difference in PFS/OS between FCR and ibrutinib + rituximab in IGHV-mutated patients.
Toxicities limit its use:
- Short-term: myelosuppression, neutropenia, infections
- Long-term: therapy-related myeloid neoplasm risk ~3–5% (almost always fatal)
BR (bendamustine + rituximab): Less intensive option; largely superseded by targeted therapies.
4. Treatment Decision Framework
New CLL diagnosis
│
▼
Asymptomatic? ──YES──► Watch & Wait (regular monitoring)
│
NO (iwCLL criteria met)
│
▼
Check: TP53/del(17p)? IGHV? FISH? Fitness/comorbidities?
│
├── del(17p) / TP53 mutation ──► BTK inhibitor OR venetoclax-based
│ (NOT chemoimmunotherapy)
│
├── IGHV mutated, fit, young ──► BTK inhibitor OR venetoclax-based
│ (FCR still an option for select pts)
│
├── IGHV unmutated ──────────► BTK inhibitor OR venetoclax-based
│
└── Unfit / elderly ─────────► BTK inhibitor (continuous) OR
VO (fixed 1 year)
5. Relapsed / Refractory CLL
| Scenario | Preferred Approach |
|---|---|
| Relapse on chemoimmunotherapy | BTK inhibitor OR venetoclax-based |
| Relapse on BTK inhibitor | Switch to venetoclax ± anti-CD20 |
| Relapse on venetoclax | BTK inhibitor OR BTKi + venetoclax combination |
| del(17p) / TP53 mutated | BTK inhibitor (zanubrutinib/acalabrutinib preferred) |
| Fit responders | Consider allogeneic SCT (only curative option) |
The 2024 ESMO Clinical Practice Guideline update (Eichhorst et al., Ann Oncol 2024; PMID 38969011) endorses BTK inhibitors and venetoclax-based combinations as frontline and relapse standards, with covalent BTK inhibitors favored due to tolerability data.
6. Special Complications — Management
Autoimmune Cytopenias
| Complication | First-Line | Second-Line |
|---|---|---|
| AIHA (warm IgG) | Glucocorticoids + transfusion | Rituximab; treat underlying CLL |
| ITP | Glucocorticoids + IVIg | Rituximab; TPO agonists (romiplostim, eltrombopag) |
| Evans syndrome | As above, combined | CLL-directed therapy |
Key: Cytopenias from autoimmune destruction (treat with immunosuppression) must be distinguished from cytopenias from marrow infiltration (treat underlying CLL).
Hypogammaglobulinemia & Infection
- IVIg indicated in patients with hypogammaglobulinemia + recurrent infections or IgG <300–500 mg/dL
- Viral prophylaxis (herpes, PCP) with most CLL therapies
- Vaccinations (live vaccines contraindicated): influenza, COVID-19, zoster (recombinant), pneumococcal, RSV
Richter's Transformation (~0.5%/year → DLBCL)
- Diagnose with FDG-PET/CT (SUV ≥10 highly suspicious) + excisional biopsy
- Treat as aggressive DLBCL (chemoimmunotherapy ± CAR-T, bispecific antibodies)
- Median survival: 6–16 months (clonally related) vs. ~5 years (clonally unrelated)
- Allogeneic SCT for responding fit patients
7. Novel & Investigational Approaches
- Non-covalent BTK inhibitors (pirtobrutinib): Active in covalent BTKi-resistant CLL (C481S mutation); approved for relapsed/refractory CLL
- CAR-T cell therapy: Promising results in small cohorts; active clinical investigation
- Bispecific antibodies: Early encouraging data in Richter's and refractory CLL
- Combination BTKi + BCL2 inhibitor: Fixed-duration IVO showing strong PFS data; may allow treatment-free intervals
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 112 — Woyach & Byrd
- ESMO Clinical Practice Guideline update — Eichhorst et al., Ann Oncol 2024 (PMID 38969011)
This is a shared conversation. Sign in to Orris to start your own chat.