Here is your complete, systematically organized MUHS 2nd MBBS Pathology answer set, sourced from Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease.
MUHS 2nd MBBS Pathology - Complete Answer Set
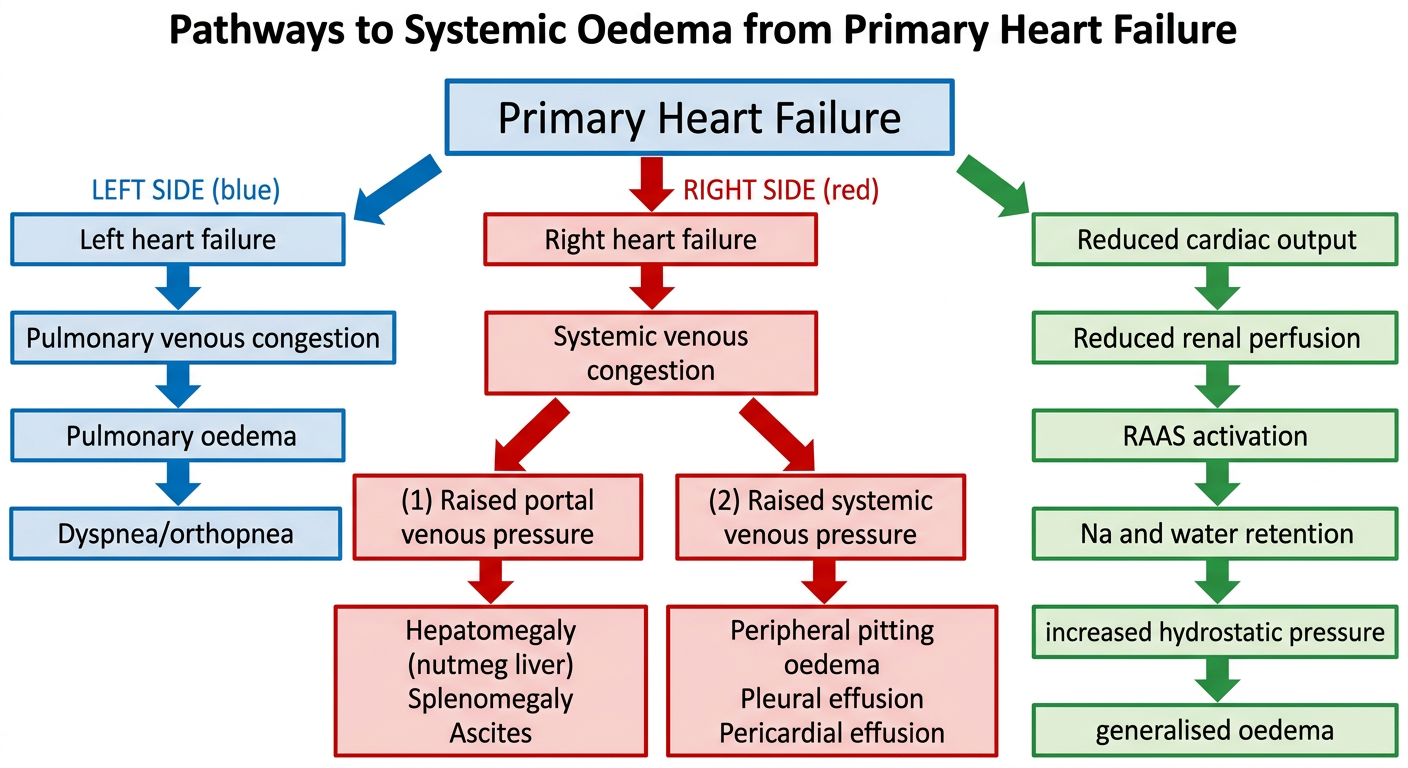
TOPIC 1: Pathways Leading to Systemic Oedema from Primary Heart Failure
Diagram:
Explanation of Pathways:
LEFT-SIDED HEART FAILURE pathway:
- Failing left ventricle → back-pressure in pulmonary veins → pulmonary capillary hypertension → transudate leaks into alveoli → pulmonary oedema
- Clinical: dyspnea, orthopnea, paroxysmal nocturnal dyspnea, fine basal crepitations
RIGHT-SIDED HEART FAILURE pathway:
- Back-pressure in systemic/portal veins → raised hydrostatic pressure
- Raised portal pressure: congestive hepatomegaly (nutmeg liver), splenomegaly, ascites
- Raised systemic venous pressure: dependent pitting oedema (ankles/feet), pleural effusion, pericardial effusion
NEUROHORMONAL/RENAL PATHWAY (common to both):
- Reduced cardiac output → reduced renal perfusion → RAAS activation → aldosterone-driven Na⁺ and H₂O retention → increased plasma volume → further raised hydrostatic pressure → generalised oedema
- ADH (vasopressin) also released, worsening fluid retention
- As heart failure worsens, retained fluid increases hydrostatic pressure and perpetuates oedema
TOPIC 2: Liver, Spleen in Right-Sided Heart Failure & Chronic Venous Congestion (CVC) of Lungs
LIVER - Right-Sided Heart Failure
Gross Features:
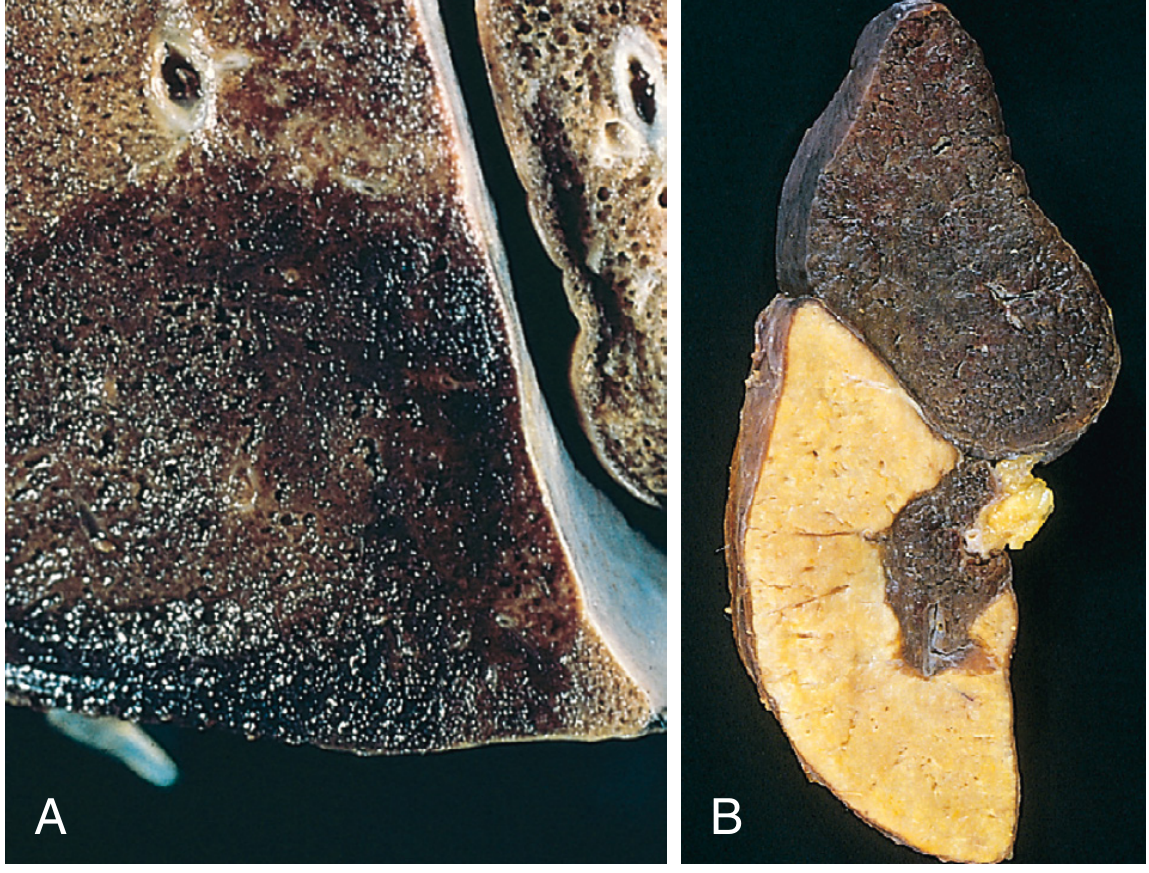
- Liver enlarged and heavy (congestive hepatomegaly)
- Cut section: dark-red congested centrilobular (central vein) zones surrounded by paler, yellowish-tan peripheral parenchyma
- This alternating pattern = "Nutmeg liver" (resembles a cut nutmeg)
- In severe/long-standing cases: central areas become fibrotic → "Cardiac cirrhosis"
Microscopic Features:
- Centrilobular sinusoidal dilatation and congestion (red cells engorging sinusoids around central vein)
- Central hepatocyte atrophy and necrosis (centrilobular necrosis) due to hypoxia - especially when left-sided failure also present
- Periportal hepatocytes survive longer (better oxygen from portal blood)
- Chronic cases: centrilobular fibrosis spreading outward → cardiac cirrhosis
SPLEEN - Right-Sided Heart Failure
Gross: Enlarged, tense, firm (congestive splenomegaly) due to raised portal venous pressure
Microscopic: Dilated sinusoids, red pulp congested with RBCs; chronic cases show fibrosis of sinusoidal walls
CHRONIC VENOUS CONGESTION (CVC) OF LUNGS
Gross Features:
- Lungs heavy, firm, brown in colour ("brown induration of lung")
- On cut section: frothy, blood-tinged fluid exudes
- Lungs 2-3 times normal weight
- Subpleural petechiae may be visible
Microscopic Features:
- Alveolar capillaries and septal vessels engorged with blood
- Alveolar walls thickened and fibrotic (congestion pneumonitis)
- Heart failure cells - alveolar macrophages laden with haemosiderin (from phagocytosed extravasated RBCs); these are pathognomonic
- Perl's Prussian blue stain demonstrates haemosiderin in macrophages
- Alveoli may contain RBCs and transudate fluid
In acute left heart failure: watery transudate floods alveoli (pulmonary oedema); in chronic CVC: the above fibrotic/haemosiderin picture predominates.
TOPIC 3: Difference Between Transudate and Exudate
| Feature | Transudate | Exudate |
|---|
| Definition | Filtration of plasma through intact capillary walls due to altered Starling forces | Fluid with high protein/cells leaking through injured/inflamed capillaries |
| Mechanism | Increased hydrostatic pressure OR decreased oncotic pressure | Increased vascular permeability (inflammation) |
| Protein content | Low (<3 g/dL) | High (>3 g/dL) |
| Specific gravity | <1.012 | >1.020 |
| Appearance | Clear, straw-coloured, translucent | Cloudy, turbid |
| Cells | Few (<1000/μL, mostly mesothelial) | Many (neutrophils, macrophages) |
| LDH | Low | High (>200 IU/L) |
| Pleural fluid:serum protein | <0.5 | >0.5 (Light's criteria) |
| Causes | CCF, nephrotic syndrome, cirrhosis, hypoalbuminaemia | Pneumonia, TB, malignancy, pancreatitis, PE |
| Fibrin | Absent or scanty | Present (may clot) |
| Glucose | Normal (serum level) | May be low (consumed by cells) |
Mnemonic: Transudate = "Thin, Transparent, sTarling forces Trouble" ; Exudate = "Extensive inflammation, Exudes protein"
TOPIC 4: Oedema
Definition
Oedema is an abnormal accumulation of interstitial fluid (water + electrolytes) in excess in the extracellular tissue spaces, body cavities, or both.
- Anasarca = generalised massive oedema involving subcutaneous tissue, serous cavities
- Hydrothorax = pleural effusion; Hydropericardium; Ascites (peritoneal)
Types
- Localised oedema - inflammatory, lymphatic, venous obstruction (DVT, tourniquet)
- Generalised oedema (anasarca) - cardiac failure, nephrotic syndrome, hepatic cirrhosis
- Pitting oedema - fluid shifts on pressure; in hypoproteinaemia, CCF
- Non-pitting oedema - lymphoedema (myxoedema), lymphatic obstruction
- Pulmonary oedema - fluid in alveoli/interstitium
- Cerebral oedema - vasogenic or cytotoxic
Pathogenesis (Starling Forces)
Normally fluid balance is maintained by:
- Forces pushing out of capillaries: hydrostatic pressure (capillary ~32 mmHg arterial end)
- Forces pulling back into capillaries: plasma colloid osmotic pressure (~25 mmHg, primarily albumin)
- Lymphatic drainage removes excess
Oedema results from imbalance:
| Mechanism | Cause | Example |
|---|
| ↑ Hydrostatic pressure | Venous obstruction, CCF | Dependent oedema in CCF |
| ↓ Plasma oncotic pressure | Hypoalbuminaemia | Nephrotic syndrome, starvation, cirrhosis |
| ↑ Vascular permeability | Inflammation (histamine, bradykinin) | Inflammatory oedema, burns |
| Lymphatic obstruction | Fibrosis, filariasis, surgery | Lymphoedema, elephantiasis |
| Na⁺/H₂O retention | RAAS activation, renal failure | Renal oedema, CCF |
Pulmonary Oedema - Pathophysiologic Mechanism
Haemodynamic (most common - CCF):
Left heart failure → pulmonary venous pressure ↑ → pulmonary capillary wedge pressure >25 mmHg → transudate floods alveoli + interstitium → impaired O₂ diffusion → hypoxia
Non-haemodynamic (ARDS, injury):
Direct alveolar/capillary injury → increased permeability → proteinaceous exudate floods alveoli
Gross:
- Heavy, wet lungs (2-3× normal weight)
- Frothy, blood-tinged fluid on cut section
- Congested, dark red appearance
Microscopic:
- Alveolar capillaries engorged
- Alveoli filled with homogeneous pink proteinaceous fluid (transudate)
- RBCs in alveolar spaces
- Heart failure cells (haemosiderin-laden macrophages) in chronic cases
- In ARDS: hyaline membrane formation lining alveolar walls
TOPIC 5: Shock
Definition
Shock is a state of systemic tissue hypoperfusion due to reduced cardiac output and/or reduced effective circulating blood volume, resulting in cellular hypoxia and, if prolonged, irreversible tissue injury and death.
(Robbins & Kumar: "a state in which diminished cardiac output or reduced effective circulating blood volume impairs tissue perfusion and leads to cellular hypoxia")
Types, Mechanisms, Examples
| Type | Mechanism | Examples |
|---|
| Cardiogenic | Pump failure → ↓ cardiac output | MI, ventricular arrhythmia, cardiac tamponade, massive PE |
| Hypovolemic | Loss of blood/plasma → ↓ preload → ↓ CO | Haemorrhage, severe burns, vomiting/diarrhoea, trauma |
| Septic | Massive inflammatory mediator release → vasodilation + vascular leakage → maldistribution of flow | Gram-positive/negative sepsis, fungal sepsis |
| Neurogenic | Loss of vasomotor tone → pooling of blood | Spinal cord injury, general anaesthesia |
| Anaphylactic | IgE-mediated hypersensitivity → systemic vasodilation + ↑ vascular permeability | Bee sting, drug allergy (penicillin) |
| Distributive | Maldistribution of perfusion (includes septic, neurogenic, anaphylactic) | As above |
| Obstructive | Mechanical obstruction to flow | Massive PE, tension pneumothorax, cardiac tamponade |
TOPIC 6: Stages of Shock
Stage 1 - NON-PROGRESSIVE (Compensated) Stage
- Baroreceptors detect ↓BP → reflex compensatory mechanisms activated
- Neurohumoral response:
- Sympathetic stimulation → tachycardia, vasoconstriction
- Catecholamine release (epinephrine, norepinephrine)
- RAAS activation → angiotensin II → aldosterone → Na⁺/H₂O retention
- ADH (vasopressin) release
- ANP counterbalances (limited)
- Effect: Peripheral vasoconstriction (skin becomes pale, cool), coronary and cerebral blood flow maintained, blood shunted to vital organs
- Clinically: Patient is compensated; vital organs perfused; reversible if treated
Stage 2 - PROGRESSIVE Stage
- If underlying cause not corrected
- Widespread tissue hypoxia → anaerobic glycolysis → lactic acidosis
- Low pH blunts arteriolar vasomotor response → arterioles dilate → blood pools in microcirculation
- Peripheral pooling → further ↓ cardiac output → endothelial ischemia → DIC
- Vital organs begin to fail
- Clinically: acidosis, oliguria (ATN), confusion, falling BP despite vasopressors
Stage 3 - IRREVERSIBLE Stage
- Severe widespread cell injury
- Lysosomal enzyme leakage → autolysis
- Myocardial contractility worsens
- Ischaemic bowel allows intestinal bacteria into bloodstream → superimposed bacteraemic shock
- Acute kidney injury (ATN) = renal failure
- Multi-organ dysfunction syndrome (MODS)
- Death despite all interventions
Morphologic Changes in Shock
- Brain: ischaemic encephalopathy
- Heart: subendocardial necrosis, contraction band necrosis
- Kidney: acute tubular necrosis (ATN) - most classic finding; shock kidney
- Lungs: diffuse alveolar damage (ARDS) - "shock lung"
- Adrenals: cortical cell lipid depletion (stress response)
- GIT: haemorrhagic gastroenteropathy
- Liver: centrilobular necrosis ("nutmeg" pattern)
Note on Hypovolemic Shock
- Caused by loss of blood (haemorrhagic) or plasma (burns, diarrhoea)
- Stages clearly apply: compensated (tachycardia, vasoconstriction, oliguria) → progressive (acidosis, falling BP) → irreversible (MOF)
- Management: aggressive fluid resuscitation (crystalloid + blood products), control the source of bleeding
- Haemorrhagic shock classified by class I-IV (% blood volume lost)
Note on Septic Shock
- Most common cause: Gram-positive bacteria (Staphylococcus, Streptococcus), then Gram-negative (E. coli, Klebsiella), fungi
- Pathogenesis: LPS (endotoxin) and other PAMPs → Toll-like receptors on macrophages/endothelium → TNF-α, IL-1, IL-6, IL-12 → massive inflammatory response
- Effects: vasodilation (warm shock initially), ↑ vascular permeability, tissue hypoperfusion, DIC, metabolic derangements
- Superantigens (TSST-1) can cause toxic shock syndrome
- Mortality: 20-30%
TOPIC 7: Thrombosis
Definition of Thrombus
A thrombus is a solid mass formed from blood constituents (platelets, fibrin, RBCs, WBCs) within the intact cardiovascular system during life (antemortem), distinguished from a postmortem clot.
Definition of Thrombosis
The process of intravascular clot (thrombus) formation within the living cardiovascular system due to activation of coagulation and/or platelet aggregation.
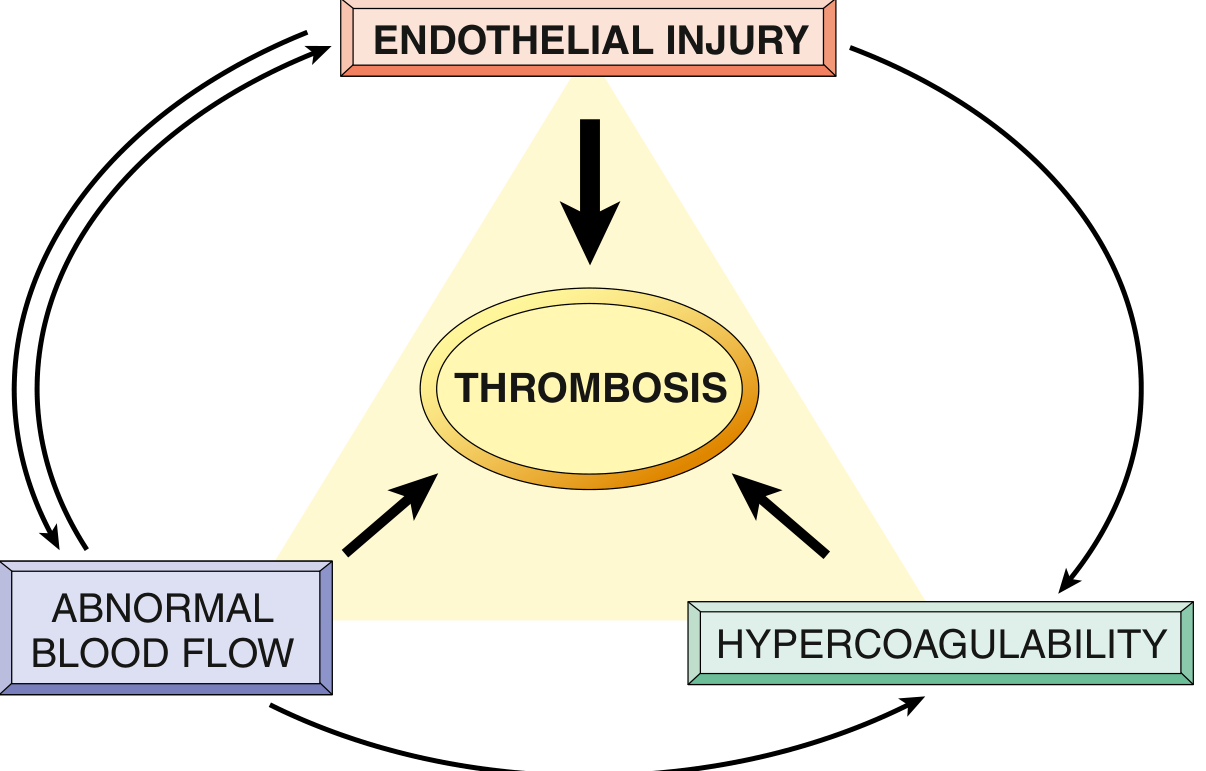
Pathogenesis - Virchow's Triad
(Robbins Fig. 3.12 - Virchow's Triad: the three interacting factors)
1. Endothelial Injury (most important)
- Exposes subendothelial collagen → platelet adhesion via vWF
- Exposes tissue factor (TF) → activates extrinsic coagulation cascade
- Endothelial activation: ↑ tissue factor, ↓ thrombomodulin, ↓ TFPI, ↑ PAI-1
- Causes: atherosclerosis, hypertension, hypercholesterolaemia, homocysteinaemia, smoking, inflammation, mechanical injury
- Predominates in arterial thrombosis
2. Abnormal (Turbulent or Stagnant) Blood Flow
- Turbulence → endothelial injury/dysfunction; causes countercurrents, local stasis; disrupts laminar flow
- Stasis → platelets contact endothelium; prevents washing away of activated clotting factors; promotes endothelial hypoxia
- Predominates in venous thrombosis
- Examples: DVT (immobility), atrial fibrillation (stasis in atrial appendage), aneurysms, atherosclerosis (turbulence at bifurcations)
3. Hypercoagulability
- Primary (genetic): Factor V Leiden mutation (most common), Prothrombin G20210A mutation, Protein C/S deficiency, Antithrombin III deficiency
- Secondary (acquired): Prolonged bed rest, pregnancy, OCP, cancer (Trousseau syndrome - migratory thrombophlebitis), antiphospholipid syndrome, nephrotic syndrome (loss of antithrombin III), HIT (heparin-induced thrombocytopenia)
Types of Thrombus
- Arterial thrombus - white/pale, platelet-rich, at sites of turbulence (coronary, cerebral arteries); often occlusive
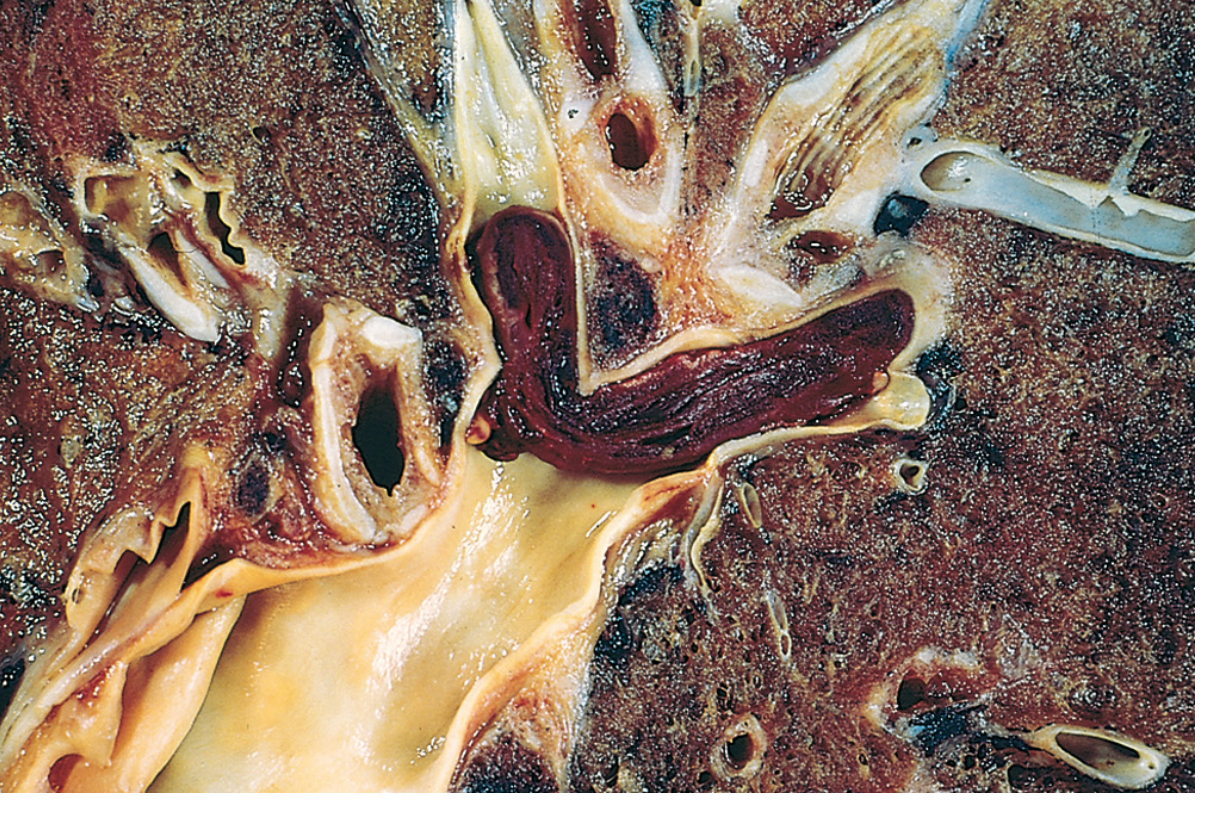
- Venous thrombus (phlebothrombosis) - red/dark, RBC-rich, at sites of stasis (lower extremity deep veins); almost always occlusive; lines of Zahn present
- Mural thrombus - in cardiac chambers or aorta (AF, MI, aneurysm); may embolise
- Valvular vegetations - on cardiac valves (infective endocarditis = infected; NBTE = sterile; Libman-Sacks = SLE)
Gross Features of Thrombus
- Focally attached to vessel wall (at point of initiation)
- Arterial thrombus: pale, grey-white, friable, platelet-rich
- Venous thrombus: dark red, gelatinous, red cell-rich
- Lines of Zahn: alternating pale (platelet + fibrin) and dark red (RBC) laminations - hallmark of antemortem thrombus
- Propagating portion: poorly attached, prone to embolisation
Microscopic Features
- Pale areas: aggregated platelets + fibrin mesh
- Dark areas: entrapped RBCs and leukocytes
- Lines of Zahn clearly visible
- Attached to vessel wall at one end
Distinguish from postmortem clot: Postmortem = gelatinous, dark red lower layer + yellow "chicken fat" upper layer, NOT attached to wall, NO lines of Zahn
Fate of Thrombus (4 possible fates)
- Propagation - thrombus extends, increasing risk of occlusion/embolism
- Embolisation - thrombus (or portion) dislodges → distant occlusion
- Dissolution (thrombolysis) - plasmin digests fibrin; most favourable outcome
- Organisation and recanalisation - ingrowth of endothelial cells, smooth muscle, fibroblasts → endothelialised channels form through the thrombus, partially restoring blood flow; becomes incorporated into vessel wall
Complications of Thrombosis
- Occlusion → ischaemia/infarction (MI, stroke, DVT-related limb ischaemia)
- Embolism → PE (from DVT), stroke (from cardiac mural thrombus)
- Cardiac mural thrombus → systemic embolism
- Phlegmasia cerulea dolens (massive DVT)
- Paradoxical embolism (venous thrombus crosses ASD/VSD to systemic circulation)
TOPIC 8: Embolism
Definition of Embolus
A detached intravascular solid, liquid, or gaseous mass that is carried by the blood from its point of origin to a distant site, where it obstructs blood flow.
Definition of Embolism
The process by which an embolus travels through the vascular system and causes obstruction at a remote site.
Types of Embolism with Examples
| Type | Origin | Common Sites of Lodgement |
|---|
| Thromboembolism (most common) | Dislodged thrombus (DVT → PE; cardiac mural thrombus → systemic) | Pulmonary arteries, cerebral arteries, mesenteric arteries |
| Fat embolism | Bone marrow fat (fractures of long bones), soft tissue trauma | Pulmonary capillaries, cerebral capillaries |
| Air/Gas embolism | IV lines, surgery, decompression sickness (divers) | Right heart, pulmonary vasculature |
| Amniotic fluid embolism | Amniotic fluid enters maternal circulation (obstetric complication) | Pulmonary vasculature |
| Cholesterol/Atheroembolism | Ruptured atherosclerotic plaque | Renal arteries, lower limb arteries |
| Tumour embolism | Fragments of tumour | Pulmonary vasculature |
| Foreign body embolism | IV drugs, catheter fragments | Pulmonary/cardiac |
| Bone marrow embolism | Trauma/CPR | Pulmonary vasculature |
| Septic embolism | Infected thrombus/vegetation | Multiple organs |
Pulmonary Thromboembolism (PTE)
Source: 70-80% from lower extremity DVT; also pelvic veins, renal veins
Pathogenesis: DVT → fragment detaches → travels through IVC → right heart → pulmonary artery
Effects depend on size:
- Massive (>60% occlusion) - sudden death, acute right heart failure (cor pulmonale), cardiovascular collapse
- Medium-sized - pulmonary haemorrhage (bronchial circulation usually prevents infarction); infarction if bronchial supply also compromised (e.g., left heart failure)
- Small/multiple - may be silent; organised into fibrous webs; recurrent emboli → pulmonary hypertension
- Saddle embolus - straddles pulmonary artery bifurcation → catastrophic
60-80% are clinically silent - small emboli resolve spontaneously; organized and incorporated into vessel wall
Morphology of pulmonary infarct:
- Haemorrhagic, wedge-shaped, with base toward pleura and apex toward the occluded vessel (red infarct due to dual blood supply + congestion)
- Microscopy: coagulative necrosis, haemorrhage, congestion
Air Embolism
- Cause: Rapid entry of air into venous system (obstetric procedures, IV injection, neck trauma, open cardiac surgery); decompression sickness (Caisson disease) - rapid ascent from high pressure → dissolved N₂ forms gas bubbles in blood and tissues
- Amount needed: >100 mL can be fatal; even 5 mL can cause death in coronary or cerebral circulation
- Effects: Air lock in right ventricle → obstructs outflow → cardiac failure; cerebral/coronary embolism
- Decompression sickness ("bends"): N₂ bubbles in muscles/joints (pain), spinal cord (paralysis), lungs (chokes), brain (cerebral symptoms)
- Treatment: Hyperbaric oxygen (recompression)
Fat Embolism
- Cause: Fractures of long bones, liposuction, severe burns, pancreatitis
- Fat globules from bone marrow enter torn venules → travel to lungs → pulmonary insufficiency
- Triad: Respiratory distress, neurological symptoms (confusion, coma), petechial rash (thrombocytopenia from fat-induced platelet aggregation)
- Microscopy: Fat globules in pulmonary capillaries (stained with Sudan IV or Oil Red O)
Amniotic Fluid Embolism
- Rare but often fatal
- Squamous cells + mucin + lanugo from amniotic fluid enter maternal pulmonary vasculature → DIC + ARDS
TOPIC 9: Infarct
Definition
An infarct is an area of ischaemic necrosis caused by occlusion of the arterial supply or the venous drainage in a tissue. The underlying lesion is usually arterial thrombosis or embolism.
Types of Infarct
| Feature | Red (Haemorrhagic) Infarct | White (Anaemic/Pale) Infarct |
|---|
| Colour | Red/dark red | Pale/white/yellow |
| Mechanism | Blood seeps into necrotic zone from collaterals or congested veins | Dense tissue prevents blood seepage |
| Occurs in | 1. Venous occlusions (testicular torsion) 2. Loose/spongy tissues (lung) 3. Dual blood supply (lung, small intestine) 4. Previously congested tissues 5. After reperfusion | Solid organs with end-arterial supply (heart, spleen, kidney) |
| Examples | Lung infarct (PE), haemorrhagic cerebral infarct, haemorrhagic bowel | MI (white eventually), splenic infarct, renal infarct |
| Shape | Wedge-shaped (base on surface) | Wedge-shaped |
Additional type: Septic infarct - when infected emboli lodge → abscess formation within the necrotic zone
Morphology of Infarcts
Gross:
- Shape: Wedge-shaped, with occluded vessel at apex and organ periphery/serosal surface at base
- Fresh (0-24 hr): Poorly defined, slightly haemorrhagic, soft
- 1-2 days: Margins better defined by a rim of hyperaemia (congestion + acute inflammation)
- Days-weeks: Pale, yellow, increasingly well-defined (in solid organs); brown/dark (in lung - haemorrhagic)
- Long-standing: Replaced by grey-white fibrous scar (contracted)
- Overlying fibrinous peritonitis/pleuritis if serosal surface involved
Microscopy:
- Main feature: Coagulative necrosis (ghost outlines of cells preserved; nuclei disappear - pyknosis → karyorrhexis → karyolysis)
- Changes take 4-12 hours to appear microscopically
- Acute inflammation at margins within hours, well-defined by 1-2 days
- Macrophages infiltrate to remove debris (4-5 days onwards)
- Granulation tissue forms (days to weeks)
- Fibrosis/scar formation (weeks to months)
- Exception: Brain → liquefactive necrosis (no coagulative necrosis)
Factors Influencing Development of Infarct
- Anatomy of blood supply - end-arterial (kidney, spleen) = more susceptible; dual supply (lung, liver) = more resistant
- Rate of occlusion - slow occlusion → collateral development → less infarction
- Tissue susceptibility to ischaemia - neurons (4-5 min), cardiac myocytes (20-30 min), fibroblasts (hours-days)
- Blood oxygenation - pre-existing anaemia/cyanosis worsens outcome
(Robbins Fig. 4.18: A = haemorrhagic wedge-shaped pulmonary infarct; B = white/pale splenic infarct)
Quick Summary Table for MUHS Revision
| Topic | Key Point |
|---|
| Oedema pathogenesis | Starling forces imbalance: ↑ hydrostatic / ↓ oncotic / ↑ permeability / lymph obstruction / Na retention |
| Pulmonary oedema micro | Alveoli flooded with pink fluid + heart failure cells (haemosiderin macrophages) |
| CVC lung | Brown induration; heart failure cells; haemosiderin; fibrosis |
| Nutmeg liver | Centrilobular congestion/necrosis → red-yellow alternating pattern |
| Transudate vs Exudate | Protein <3 vs >3; SG <1.012 vs >1.020; clear vs cloudy |
| Virchow's Triad | Endothelial injury + Abnormal flow + Hypercoagulability |
| Lines of Zahn | Alternating platelets/fibrin + RBCs = antemortem thrombus |
| Fate of thrombus | Propagate, Embolise, Dissolve, Organise/Recanalise |
| Shock stages | Nonprogressive (compensated) → Progressive (acidosis, hypoxia) → Irreversible (MOF) |
| Infarct types | Red (loose tissue/dual supply/venous) vs White (solid organ/end-arterial) |
| Infarct microscopy | Coagulative necrosis (except brain = liquefactive) |
| PE silent % | 60-80% clinically silent |
| Air embolism | >100 mL fatal; decompression sickness = N₂ bubbles; treat with hyperbaric O₂ |
Sources: Robbins & Kumar Basic Pathology (10th ed.) and Robbins, Cotran & Kumar Pathologic Basis of Disease. All textbook content retrieved from the medical library database.