Von willebrand disease
Reading File
Finding Sources
Searching PubMed
"von Willebrand disease"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile a thorough response.
Von Willebrand Disease (VWD)
Definition & Epidemiology
Von Willebrand disease is the most common inherited bleeding disorder, affecting approximately 1% of the population, with clinically significant bleeding symptoms in ~1 in 1,000 individuals. Prevalence is consistent across races and ethnicities. It is caused by quantitative or qualitative deficiency of von Willebrand factor (VWF) - a large multimeric plasma glycoprotein essential for normal hemostasis.
Clinical disease is more prevalent in females despite autosomal inheritance, because they face greater hemostatic challenges (menorrhagia occurs in up to 80% of women with VWD).
- Goldman-Cecil Medicine, p. 1400-1401
- Robbins & Kumar Basic Pathology, p. 430
Structure & Function of VWF
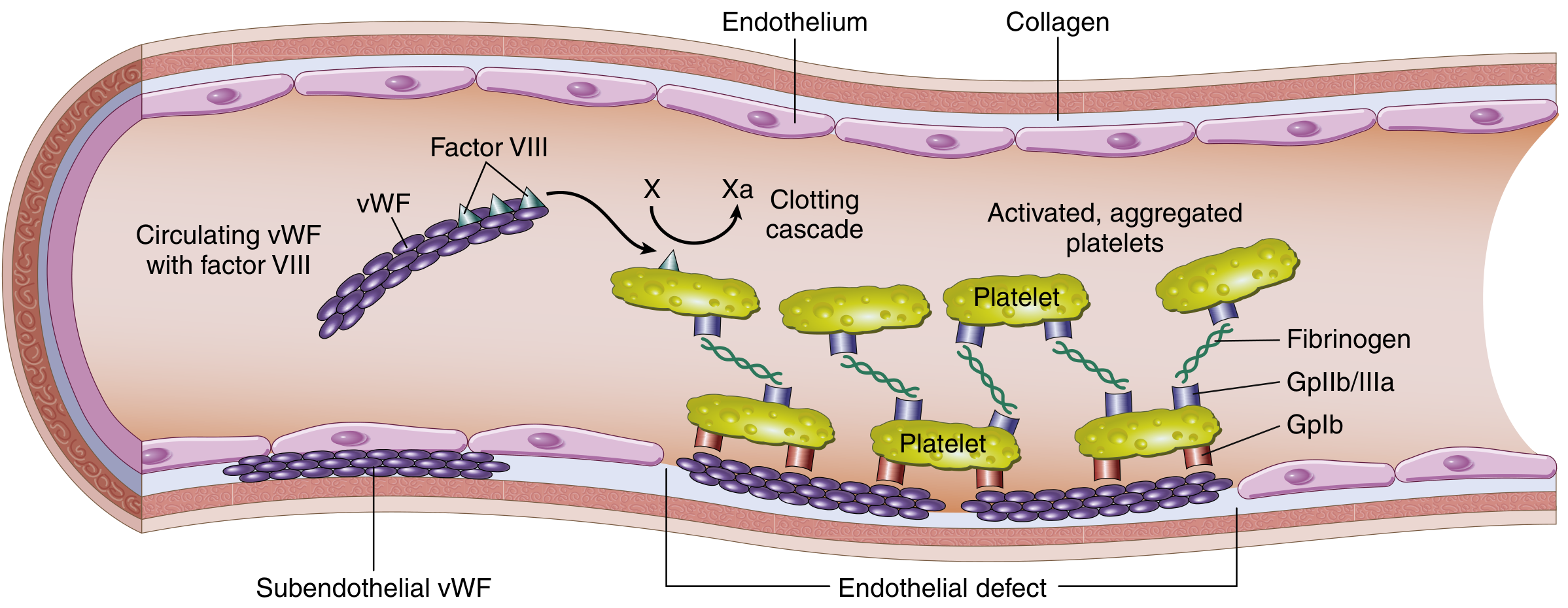
Structure and function of factor VIII-vWF complex (Robbins & Kumar)
VWF has two critical roles in hemostasis:
- Platelet adhesion - mediates platelet binding to exposed subendothelial collagen at vascular injury sites via the glycoprotein Ib (GpIb) receptor
- Factor VIII carrier - protects factor VIII from premature proteolytic degradation; the VWF-FVIII complex circulates together in plasma
Synthesis and storage:
- Produced in endothelial cells and megakaryocytes
- Stored in Weibel-Palade bodies (endothelium) and alpha-granules (platelets)
- Released by secretagogues: histamine, thrombin, DDAVP
- VWF monomers dimerize in the endoplasmic reticulum, then form high-molecular-weight (HMW) multimers in the Golgi via N-terminal disulfide bonds
Multimeric structure is critical: HMW multimers bind collagen and platelets with highest affinity and are most efficient in inducing platelet aggregation under high-shear conditions.
ADAMTS13 (a metalloprotease) cleaves ultralarge VWF multimers released from endothelial cells to generate the spectrum of circulating multimers. Deficiency of ADAMTS13 leads to TTP (distinct from VWD).
- Goldman-Cecil Medicine, p. 1405-1413
- Tietz Textbook of Laboratory Medicine 7th Ed, p. 3073
Classification and Subtypes
| Type | Description | FVIII | VWF:RCo | VWF:Ag | HMW Multimers |
|---|---|---|---|---|---|
| 1 | Quantitative deficiency (most common, ~70-80%) | ↓ or N | <30% | <30% | Normal distribution, decreased quantity |
| 2A | Qualitative - loss of HMW multimers (not synthesized) | ↓ or N | ↓ | N to ↓ | Absent |
| 2B | Qualitative - "hyperfunctional" HMW multimers rapidly cleared; spontaneous platelet aggregation; mild thrombocytopenia | ↓ or N | ↓ | N to ↓ | Absent (↑ RIPA at low dose ristocetin) |
| 2M | Qualitative defect - decreased platelet binding without multimer loss | ↓ or N | ↓ | N to ↓ | Normal |
| 2N | Qualitative defect - only in FVIII binding (mimics hemophilia A) | ↓↓ | Normal | Normal | Normal |
| 3 | Near-complete absence of VWF (most severe, autosomal recessive) | ↓↓ | <3% | <3% | Absent |
| "Low VWF" | Borderline values, often not true hereditary VWD | N | 30-50% | 30-50% | Normal |
- Goldman-Cecil Medicine, Table 59-1
Genetics:
- Types 1, 2, 3: autosomal
- Type 1: autosomal dominant, reduced quantity of VWF
- Type 3: autosomal recessive (homozygous), near-total absence - can cause features resembling hemophilia due to severe FVIII deficiency
- The VWF gene spans 178 kb on chromosome 12, comprising 52 exons
Clinical Manifestations
Mucocutaneous bleeding pattern (distinguishes VWD from hemophilias):
- Epistaxis
- Gum bleeding
- Easy bruising
- Prolonged bleeding from minor wounds
- Menorrhagia (hallmark in women; up to 80% of female patients)
- Recurrent gastrointestinal bleeding (especially types 2 and 3)
In rare homozygous (type 3) patients: deep tissue bleeds and hemarthroses can occur due to concomitant severe FVIII deficiency, resembling hemophilia.
Diagnosis
Initial Labs
| Test | Purpose |
|---|---|
| VWF antigen (VWF:Ag) | Quantifies total VWF protein |
| VWF ristocetin cofactor activity (VWF:RCo) | Assesses functional VWF |
| Factor VIII activity | Reduced in severe VWD |
| RCo:Ag ratio | <0.7 suggests qualitative defect (type 2) |
Normal plasma VWF: 50-150 IU/dL. Blood group O individuals have ~25% lower levels than group AB.
Supplementary Tests (for type 2 subclassification)
- VWF multimer analysis - electrophoresis to assess multimer distribution; required for 2A, 2B, 2M
- RIPA (ristocetin-induced platelet aggregation) - abnormal response to low-dose ristocetin (0.5 mg/mL) is diagnostic for type 2B
- VWF:FVIII binding assay - low binding capacity diagnoses type 2N (mimics hemophilia A)
- Tietz Textbook of Laboratory Medicine 7th Ed, p. 4092-4101
Treatment
1. DDAVP (Desmopressin)
Drug of choice for type 1 VWD and some type 2A/2M patients.
- Mechanism: releases VWF from Weibel-Palade bodies, transiently elevating plasma VWF
- Routes:
- IV: 0.3 µg/kg in 100 mL normal saline over 20 minutes (max 25-30 µg)
- Intranasal: 300 µg total (or 150 µg per nostril for persons >50 kg)
- Contraindicated in type 2B (releases abnormal HMW multimers that worsen thrombocytopenia)
- Tachyphylaxis occurs with repeated use; response decreases after 2-3 doses (Weibel-Palade stores deplete)
- Test dose recommended before elective use
2. VWF Concentrate (Factor Replacement)
For major surgery, severe bleeds, type 3, or DDAVP-unresponsive patients:
- Initial dose: 40-60 RCo units/kg; Maintenance: 20-40 RCo units/kg every 8-24 hr
- Monitor trough and peak FVIII and RCo daily
- Targets: troughs >50% RCo; peaks <200% RCo, <250% FVIII
3. Antifibrinolytics
Tranexamic acid (oral 25 mg/kg three times daily or IV 15 mg/kg three times daily):
- Useful adjunct for mucosal bleeding, dental procedures (7-10 day course)
- Minimal thrombotic risk even in high-risk populations
- Avoid with significant hematuria (risk of clot retention and urinary obstruction)
4. Hormonal therapy
Combined oral contraceptives for menorrhagia in women.
Acquired von Willebrand Syndrome
VWD can be acquired (not inherited), with no personal or family history of lifelong bleeding. Causes include:
- Autoantibodies - lymphoproliferative disorders, myeloma, MGUS
- VWF adsorption onto cells - Wilms tumor (expressing GpIb), myeloproliferative disorders with elevated platelets
- Increased shear-force proteolysis - aortic stenosis, LVAD, congenital heart disease (Heyde syndrome)
- Hypothyroidism - decreased VWF synthesis/secretion
- Tietz Textbook of Laboratory Medicine 7th Ed, p. 4104-4118
Key Distinguishing Points
| Feature | VWD | Hemophilia A |
|---|---|---|
| Pattern | Mucocutaneous | Deep tissue/hemarthroses |
| Inheritance | Autosomal | X-linked recessive |
| Sex affected | Both (more symptomatic in females) | Predominantly males |
| vWF | Reduced or dysfunctional | Normal |
| FVIII | Normal to mildly reduced | Markedly reduced |
| Platelet count | Usually normal (low in type 2B) | Normal |
| BT/PFA-100 | Prolonged | Normal |
Sources: Goldman-Cecil Medicine (International Edition), p. 1400-1575 | Robbins & Kumar Basic Pathology, p. 430-431 | Tietz Textbook of Laboratory Medicine 7th Ed, p. 3073-4140
FNAC
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the complete FNAC overview.
Fine Needle Aspiration Cytology (FNAC)
Definition & Overview
FNAC is a minimally invasive diagnostic technique that uses a thin needle (typically 22-27 gauge) to aspirate cells from a mass lesion for cytological examination. It is widely available, simple, relatively safe, and can be performed as an outpatient procedure with little patient discomfort.
- Bailey and Love's Short Practice of Surgery 28th Ed
- Cummings Otolaryngology Head & Neck Surgery
Principle of FNAC
The needle is inserted into the target lesion, and negative pressure (suction) is applied via a syringe to aspirate cells. The aspirated material is then smeared onto glass slides, fixed (wet or air-dried), and stained for cytological analysis. It yields individual cells and cell clusters but not tissue architecture - this is the key limitation distinguishing it from core needle biopsy and histopathology.
Technique
Equipment:
- 22-25 gauge needle (fine needle), 10-20 mL syringe, syringe holder/pistol grip
- Glass slides, fixative (95% ethanol for Papanicolaou stain; air-dry for Giemsa/Diff-Quik)
Steps:
- Palpate and immobilize the lesion with the non-dominant hand
- Clean the skin with antiseptic; local anaesthesia usually not required for superficial lesions
- Insert the needle into the lesion
- Apply suction by withdrawing the syringe plunger (negative pressure)
- Make multiple passes through the lesion while maintaining suction (fan-shaped movements)
- Release suction before withdrawing the needle (to prevent aspirate entering the barrel)
- Express material onto glass slides and smear immediately
- Fix and stain
Ultrasound guidance is strongly recommended - it improves sensitivity, specificity, and accuracy, particularly for non-palpable, deep, or complex lesions.
Non-aspiration (capillary) technique - used for hypervascular lesions (e.g., thyroid) to reduce blood contamination; relies on capillary action alone without syringe suction.
Advantages
| Advantage | Detail |
|---|---|
| Minimally invasive | No incision required; outpatient procedure |
| Rapid results | Same-day diagnosis possible with on-site evaluation |
| High accuracy | Sensitivity 80-99%, specificity 97-100% depending on site |
| Cost-effective | Significantly reduces unnecessary surgeries |
| Safe | Very low complication rate vs. larger bore biopsies |
| Diagnostic AND therapeutic | Can drain cysts (breast, thyroid) while sampling |
Limitations
-
Cannot assess tissue architecture - cannot distinguish invasive from in-situ cancer, or follicular adenoma from follicular carcinoma (requires capsular/vascular invasion assessment)
-
Operator dependent - accuracy varies greatly with aspirator skill and cytopathologist experience
-
Sampling error - inadequate/non-diagnostic rate ~5-15%; false-negative rate 1-6%
-
Ancillary studies limited - lymphoma and high-grade malignancies require flow cytometry and immunohistochemistry, which need core needle biopsy for adequate tissue
-
Non-diagnostic results should never be interpreted as negative for malignancy
-
Mulholland and Greenfield's Surgery 7th Ed, p. 3883
-
Bailey and Love's 28th Ed
Applications by Site
1. Thyroid Nodules (Primary indication)
FNAC is the single most accurate and cost-effective investigative procedure for thyroid nodules. It has:
- Decreased the number of patients requiring surgery by 20-75%
- Nearly tripled the yield of carcinomas in surgical specimens
- Sensitivity >80%, specificity >90%
Indications for FNA (based on ultrasound features and size):
- High/intermediate suspicion features (solid hypoechoic with microcalcifications): nodule ≥1 cm
- Low suspicion features: nodule ≥1.5 cm
- Very low suspicion (spongiform/partially cystic): nodule ≥2.0 cm
- Purely cystic nodules: FNA generally not recommended diagnostically (can be done therapeutically)
- 18FDG-PET hypermetabolic nodules: biopsy if ≥1 cm (~35% malignancy risk)
Key limitation: Follicular neoplasm vs. follicular carcinoma CANNOT be differentiated by FNAC - requires histological evidence of capsular or vascular invasion.
Bethesda System for Reporting Thyroid Cytopathology (BSRTC) - 2023 (3rd edition):
| Bethesda Category | Description | Malignancy Risk | Management |
|---|---|---|---|
| I | Nondiagnostic / Unsatisfactory | Variable | Repeat US-guided FNA |
| II | Benign | ~3% | Clinical follow-up |
| III | Atypia of Undetermined Significance (AUS) | 10-40% | Repeat FNA or molecular testing |
| IV | Follicular Neoplasm (FN) | 20-40% | Lobectomy vs. molecular testing |
| V | Suspicious for Malignancy | ~75% | Near-total thyroidectomy or lobectomy |
| VI | Malignant | ~99% (98% PTC) | Surgery |
- Sabiston Textbook of Surgery, p. 1505-1506
- K.J. Lee's Essential Otolaryngology
- Goldman-Cecil Medicine
2. Salivary Gland Lesions
Performance: Sensitivity 85.5-99%, specificity 96.3-100%. Accuracy is higher for benign lesions. False-negative for malignancy occurs in up to 5% of cases.
Milan System for Reporting Salivary Gland Cytopathology (MSRSGP):
| Category | Risk of Malignancy | Management |
|---|---|---|
| I Non-diagnostic | 25% | Repeat FNAC / radiological correlation |
| II Non-neoplastic | 10% | Clinical follow-up |
| III AUS (Atypia of Undetermined Significance) | 20% | Repeat FNAC or surgery |
| IVA Benign Neoplasm | <5% | Conservative surgery or follow-up |
| IVB SUMP (Salivary gland neoplasm of Uncertain Malignant Potential) | 35% | Conservative surgery |
| V Suspicious for Malignancy | 60% | Surgery |
| VI Malignant | >90% | Surgery (extent per type/grade) |
- Bailey and Love's 28th Ed, Table 54.4
- Cummings Otolaryngology
3. Breast Lesions
FNAC is the least invasive breast biopsy method. Can be done by palpation or image guidance.
- Sensitivity 98%, specificity 97% (in experienced hands)
- Classic Triple Test (clinical + imaging + FNAC) - all three must agree for definitive benign diagnosis
- Limitation: Cannot diagnose invasive cancer (no tissue architecture) - cannot confirm invasion
- Currently, high-suspicion lesions are preferred to undergo core needle biopsy for superior histological diagnosis
- Simple breast cysts: therapeutic aspiration is a primary indication
4. Head & Neck Lymph Nodes
- Evaluation of cervical lymphadenopathy (reactive vs. metastatic vs. lymphoma)
- Avoids open biopsy in many cases
- With immunohistochemical staining for EBV RNA (EBER), diagnosis of nasopharyngeal carcinoma (NPC) with neck node metastasis can be confirmed
- For lymphoma: core needle biopsy preferred over FNAC as flow cytometry and immunohistochemistry require more tissue
5. EUS-FNA (Endoscopic Ultrasound-Guided FNA)
Used for:
-
Pancreatic masses and cysts
-
Mediastinal lymph nodes
-
Submucosal GI lesions
-
Peri-intestinal masses
-
Clinical Gastrointestinal Endoscopy 3rd Ed
6. Other Sites
| Site | Application |
|---|---|
| Liver | Focal hepatic lesions |
| Lung | Peripheral and central masses (CT-guided) |
| Kidney | Renal masses |
| Bone | Osteolytic lesions |
| Soft tissue | Subcutaneous/deep soft tissue masses |
| Mycetoma | Distinguishes eumycetoma vs. actinomycetoma |
Complications
Generally very few and minor:
- Local hematoma / bruising - most common
- Infection - rare
- Pneumothorax - with lung or deep neck FNAC
- Tumor seeding along needle track - extremely rare; theoretical concern
- Vasovagal syncope
- Inadequate sample (5-15%) - not a complication per se but a procedural failure
FNAC vs. Core Needle Biopsy vs. Excision Biopsy
| Feature | FNAC | Core Needle Biopsy | Excision Biopsy |
|---|---|---|---|
| Needle size | 22-27G (fine) | 14-18G (large bore) | Surgical |
| Yield | Cytology (cells only) | Histology (tissue cores) | Histology (whole specimen) |
| Tissue architecture | No | Yes | Yes |
| Invasion assessment | No | Yes | Yes |
| Ancillary tests (IHC, flow) | Limited | Yes | Yes |
| Complication risk | Very low | Low-moderate | Higher |
| Cost | Low | Moderate | High |
| Best for | Thyroid, salivary, cysts | Breast, lymphoma | Uncertain/failed biopsy |
Reporting Systems Summary
| Site | Reporting System |
|---|---|
| Thyroid | Bethesda System (BSRTC) - 6 categories (2023, 3rd ed.) |
| Salivary Gland | Milan System (MSRSGP) - 6 categories |
| Breast | B-classification (B1-B5) or ACR BI-RADS correlations |
| Pancreas | Papanicolaou Society guidelines |
Sources: Sabiston Textbook of Surgery (Elsevier) | Bailey & Love's Short Practice of Surgery 28th Ed | Cummings Otolaryngology 7th Ed | Mulholland & Greenfield's Surgery 7th Ed | K.J. Lee's Essential Otolaryngology | Goldman-Cecil Medicine (International Ed)
This is a shared conversation. Sign in to Orris to start your own chat.