Here is your comprehensive pathology assignment on leukemia, written for pharmacy students:
LEUKEMIA — A Pathology Assignment for Pharmacy Students
Subject: Pathology | Level: B.Pharm / Pharm.D
Reference Texts: Robbins & Cotran Pathologic Basis of Disease (10e); Robbins & Kumar Basic Pathology; Katzung's Basic and Clinical Pharmacology (16e)
1. INTRODUCTION AND DEFINITION
Leukemia refers to a group of malignant neoplasms of hematopoietic stem cells or their progenitors that primarily involve the bone marrow and peripheral blood. Unlike lymphomas, which form solid tumor masses in lymphoid organs, leukemias characteristically flood the bloodstream with abnormal (neoplastic) white blood cells. These malignant cells crowd out normal hematopoietic elements, leading to anemia, thrombocytopenia, and immunosuppression.
The term leukemia derives from the Greek for "white blood," reflecting the dramatic leukocytosis that can be observed in some forms.
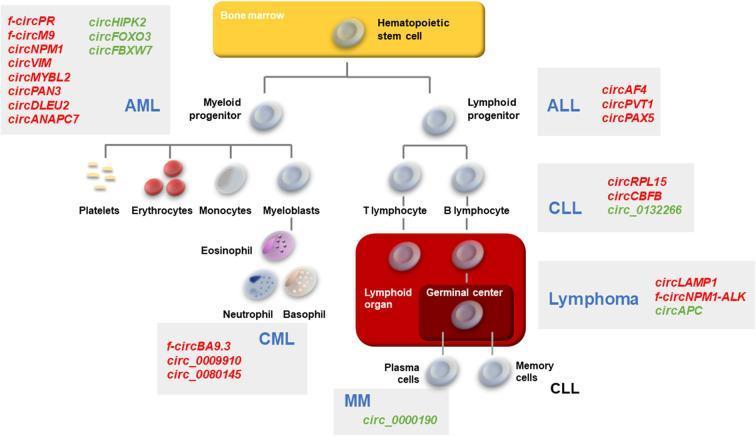
2. CLASSIFICATION
Leukemias are classified by two major criteria:
| Criterion | Categories |
|---|
| Cell lineage | Myeloid vs. Lymphoid |
| Clinical course | Acute (rapid onset, immature blasts) vs. Chronic (slower, more differentiated cells) |
This gives four principal types:
| Type | Abbreviation | Cell of Origin |
|---|
| Acute Myeloid Leukemia | AML | Myeloid progenitor |
| Chronic Myeloid Leukemia | CML | Pluripotent hematopoietic stem cell |
| Acute Lymphoblastic Leukemia | ALL | Immature B or T lymphoblast |
| Chronic Lymphocytic Leukemia | CLL | Mature (naïve) B lymphocyte |
The diagram below summarizes the hematopoietic lineage and the point of origin of each major leukemia:
3. ACUTE LYMPHOBLASTIC LEUKEMIA (ALL)
3.1 Definition
ALL is a neoplasm composed of immature B (pre-B) or T (pre-T) lymphocytes called lymphoblasts. About 85% are B-cell ALL (B-ALL), which typically manifests as childhood acute leukemia; the remaining 15% are T-cell ALL (T-ALL), which tends to present in adolescent males, often as a thymic mass.
3.2 Epidemiology
- Most common cancer of children — approximately 2,500 new cases diagnosed annually in the United States
- B-ALL peaks at ~3 years of age; T-ALL peaks in adolescence
- Slightly more frequent in boys than girls
- Hispanic/Latino children have the highest incidence of ALL among ethnic groups in the United States
- Both B-ALL and T-ALL also occur, less commonly, in adults
3.3 Pathogenesis & Molecular Mechanisms
ALL arises through accumulation of mutations that:
- Dysregulate transcription factors required for normal lymphoid development, causing maturation arrest and increased self-renewal
- Activate kinase signaling (e.g., tyrosine kinase mutations, RAS signaling) to promote proliferation
Key mutations and cytogenetic abnormalities:
| Mutation / Translocation | Impact |
|---|
| NOTCH1 mutations | Present in most T-ALLs; NOTCH1 is essential for T-cell development |
| PAX5, TCF3, ETV6, RUNX1, BCR::ABL1, KMT2A, PBX1 mutations | Present in B-ALL; required for early hematopoietic differentiation |
| t(12;21) — ETV6::RUNX1 | Most common translocation in B-ALL (~25%); excellent prognosis |
| t(9;22) — BCR::ABL1 (Philadelphia chromosome) | Present in ~25% of adult B-ALL; poor prognosis; responsive to TKIs |
| Hyperdiploidy (>50 chromosomes) | Favorable prognosis in B-ALL |
| Hypodiploidy (<44 chromosomes) | Poor prognosis |
| t(4;11) — KMT2A rearrangement | Seen in infant B-ALL and adult ALL; very poor prognosis |
| CDKN2A deletions | Frequent in T-ALL |
| TAL1 rearrangements | Activate oncogenic transcription factors in T-ALL |
Mutations in transcription factor genes alone are insufficient for ALL — additional driver mutations promoting growth are necessary, consistent with the multistep model of carcinogenesis.
3.4 Morphology
- Peripheral blood: Leukocytosis with lymphoblasts; cytopenias (anemia, thrombocytopenia) in some cases
- Bone marrow: Hypercellular; diffuse infiltration by lymphoblasts — medium-sized cells with scant cytoplasm, dispersed chromatin, and inconspicuous nucleoli
- Lymphoblasts are PAS-positive (contain cytoplasmic glycogen)
- Absent Auer rods (key distinguishing feature from AML)
3.5 Immunophenotype
- B-ALL: TdT+, CD19+, CD10+ (CALLA), CD20± , surface Ig−
- T-ALL: TdT+, CD2+, CD3+, CD5+, CD7+
- TdT (terminal deoxynucleotidyl transferase) is a nuclear enzyme expressed in immature lymphoblasts — a key diagnostic marker
3.6 Clinical Features
- Abrupt onset of symptoms reflecting bone marrow failure:
- Fatigue, pallor (anemia)
- Fever, infections (neutropenia)
- Bleeding, petechiae (thrombocytopenia)
- Bone pain (marrow infiltration)
- Lymphadenopathy, hepatosplenomegaly
- CNS involvement — headache, vomiting (occurs in a minority but critical to treat/prevent)
- T-ALL: superior mediastinal mass (thymic enlargement) — may cause superior vena cava syndrome
3.7 Laboratory Findings
- Peripheral blood: elevated WBC (lymphoblasts), decreased RBC and platelets
- LDH elevated (reflects rapid cell turnover)
- Uric acid elevated (hyperuricemia — risk of tumor lysis syndrome)
- Bone marrow biopsy: >20% blasts (WHO criterion for acute leukemia)
3.8 Treatment & Pharmacology
Treatment follows three phases: Induction → Consolidation → Maintenance
| Phase | Drug Classes / Agents |
|---|
| Induction | Vincristine, glucocorticoids (prednisolone/dexamethasone), L-asparaginase, anthracyclines (daunorubicin) |
| Consolidation | High-dose methotrexate, cytarabine, 6-mercaptopurine |
| Maintenance | Oral 6-mercaptopurine + weekly methotrexate (2–3 years) |
| CNS prophylaxis | Intrathecal methotrexate ± cytarabine |
| Ph+ ALL (BCR::ABL1) | Add tyrosine kinase inhibitor: imatinib or dasatinib |
| Relapsed/Refractory | Blinatumomab (anti-CD19/CD3 BiTE), inotuzumab ozogamicin (anti-CD22), CAR-T cells |
3.9 Prognosis
- Children with B-ALL: ~90% cure rate with modern chemotherapy (one of the great successes of oncology)
- Adults: 5-year survival ~40%; worse prognosis
- Favorable factors: age 1–10 years, hyperdiploidy, ETV6::RUNX1 fusion, low WBC at diagnosis
- Unfavorable factors: Philadelphia chromosome (t(9;22)), KMT2A rearrangement, hypodiploidy, adult age, T-ALL with complex cytogenetics
4. ACUTE MYELOID LEUKEMIA (AML)
4.1 Definition
AML is a clonal neoplasm of myeloid progenitor cells characterized by the accumulation of immature blasts (myeloblasts) in the bone marrow and blood, with failure of normal hematopoiesis.
4.2 Epidemiology
- Most common acute leukemia in adults; median age at diagnosis ~65 years
- Accounts for ~80% of acute leukemias in adults
- Incidence increases with age
- Slightly more common in males
4.3 Etiology and Risk Factors
- Prior myelodysplastic syndrome (MDS) or myeloproliferative neoplasm ("secondary AML")
- Chemotherapy / radiation exposure — alkylating agents, topoisomerase II inhibitors (e.g., etoposide)
- Occupational exposure to benzene
- Genetic predisposition: Down syndrome (trisomy 21) — markedly increased risk in children; Fanconi anemia; Bloom syndrome
4.4 Pathogenesis & Molecular Mechanisms
AML involves two classes of cooperating mutations:
- Class I mutations — activate tyrosine kinase/RAS signaling → promote proliferation (e.g., FLT3-ITD, RAS mutations)
- Class II mutations — disrupt transcription factors → block differentiation / maturation arrest (e.g., PML::RARA, RUNX1::RUNX1T1, CBFβ::MYH11)
Key translocations and mutations:
| Genetic Abnormality | Associated AML Subtype | Clinical Significance |
|---|
| t(15;17) — PML::RARA | Acute Promyelocytic Leukemia (APL, AML-M3) | Sensitive to ATRA + arsenic trioxide; risk of DIC |
| t(8;21) — RUNX1::RUNX1T1 | AML with maturation (M2) | Favorable prognosis |
| inv(16) — CBFβ::MYH11 | Acute myelomonocytic leukemia with eosinophilia (M4Eo) | Favorable prognosis |
| FLT3-ITD | Various; de novo AML | Poor prognosis; FLT3 inhibitors (midostaurin, gilteritinib) available |
| NPM1 mutation | De novo AML | Favorable if without FLT3-ITD |
| TP53 mutation | Therapy-related AML | Very poor prognosis |
| IDH1/IDH2 mutations | Various | Targeted therapy: enasidenib (IDH2), ivosidenib (IDH1) |
4.5 Morphology
- Peripheral blood: Myeloblasts with prominent nucleoli and Auer rods
- Auer rods — pink, needle-shaped cytoplasmic inclusions of abnormal primary granules; pathognomonic for AML
- Bone marrow: Markedly hypercellular; >20% myeloblasts (WHO criterion); near-complete replacement of normal marrow
- Myeloperoxidase (MPO) stain positive in myeloid lineage cells
4.6 FAB Classification (Older, Still Clinically Referenced)
| FAB Class | Name | Distinctive Feature |
|---|
| M0 | Minimally differentiated AML | No granules/Auer rods |
| M1 | AML without maturation | Myeloblasts without differentiation |
| M2 | AML with maturation | t(8;21) common |
| M3 | Acute promyelocytic leukemia (APL) | Auer rods in bundles ("faggot cells"); t(15;17) |
| M4 | Acute myelomonocytic leukemia | Both myeloid and monocytic differentiation |
| M4Eo | As above + abnormal eosinophils | inv(16) |
| M5 | Acute monocytic leukemia | Gum infiltration common |
| M6 | Erythroleukemia | Predominant erythroid precursors |
| M7 | Acute megakaryoblastic leukemia | Myelofibrosis; Down syndrome association |
4.7 Clinical Features
- Abrupt onset of fatigue, pallor (anemia)
- Fever, recurrent infections (neutropenia)
- Bleeding, bruising, petechiae (thrombocytopenia)
- APL (M3): Life-threatening disseminated intravascular coagulation (DIC) due to release of procoagulant substances from granules
- Monocytic AML (M4/M5): Gingival hyperplasia, skin infiltration (leukemia cutis)
- Hepatosplenomegaly (less pronounced than in CML)
- Chloroma (myeloid sarcoma): Extramedullary mass of myeloid blasts, may appear greenish due to MPO
4.8 Treatment & Pharmacology
| Phase | Drug Classes / Agents |
|---|
| Standard induction ("7+3" regimen) | Cytarabine (7 days continuous infusion) + anthracycline (daunorubicin or idarubicin, 3 days) |
| APL (M3) — first-line | All-trans retinoic acid (ATRA) + arsenic trioxide (ATO) — achieves differentiation of malignant promyelocytes |
| Targeted therapy — FLT3-ITD | Midostaurin (added to induction), gilteritinib (relapsed/refractory) |
| IDH2 mutation | Enasidenib |
| IDH1 mutation | Ivosidenib |
| Consolidation | High-dose cytarabine; allogeneic stem cell transplant for high-risk AML |
| Elderly/unfit patients | Venetoclax (BCL-2 inhibitor) + azacitidine or decitabine (hypomethylating agents) |
Pharmacy note: ATRA is a vitamin A derivative (retinoid) that acts on the PML-RARA fusion protein to restore differentiation — a landmark example of targeted, mechanism-based therapy.
4.9 Prognosis
- Without treatment: fatal within weeks to months
- With therapy: 5-year overall survival ~25–30% in adults; significantly better with favorable cytogenetics
- APL treated with ATRA + ATO: ~90% complete remission, ~70% cure
- Favorable: t(8;21), inv(16), NPM1 mutation without FLT3-ITD
- Unfavorable: TP53, complex karyotype, therapy-related AML, FLT3-ITD
5. CHRONIC MYELOID LEUKEMIA (CML)
5.1 Definition
CML is a myeloproliferative neoplasm characterized by the presence of a chimeric BCR::ABL1 gene, which encodes a constitutively active tyrosine kinase. It is distinguished from other myeloproliferative neoplasms by this specific genetic abnormality.
5.2 Molecular Basis — The Philadelphia Chromosome
- In >90% of cases, BCR::ABL1 arises from a reciprocal t(9;22)(q34;q11) translocation — the Philadelphia chromosome (Ph)
- The fusion produces a 210 kDa BCR-ABL tyrosine kinase (p210)
- This kinase is constitutively active → drives uncontrolled myeloid proliferation
- Cell of origin: pluripotent hematopoietic stem cell
- In remaining cases (~10%), the fusion is formed by cryptic rearrangements detectable only by FISH or PCR
5.3 Epidemiology
- Predominantly a disease of adults (median age 50–60 years)
- Rare in children (<10% of childhood leukemias)
- Slight male predominance
5.4 Pathogenesis
BCR-ABL activates multiple downstream signaling cascades:
- RAS/MAPK pathway → cell proliferation
- PI3K/AKT pathway → cell survival, apoptosis resistance
- JAK-STAT pathway → cytokine-independent growth
The result is a massive clonal expansion of myeloid cells at all stages of maturation (not just blasts).
5.5 Natural History — Three Phases
| Phase | Features |
|---|
| Chronic phase | Lasts 3–5 years; hypercellular marrow; <10% blasts; manageable with TKIs |
| Accelerated phase | Increasing blasts (10–20%); progressive disease; cytogenetic evolution |
| Blast crisis | >20% blasts; resembles acute leukemia (70% myeloid, 30% lymphoid blast crisis); fatal without treatment |
5.6 Morphology
- Peripheral blood: Marked leukocytosis (often 50,000–200,000 cells/μL); full spectrum of myeloid cells — neutrophils, bands, metamyelocytes, myelocytes, occasional myeloblasts; basophilia is characteristic
- Bone marrow: Hypercellular (90–100% cellularity); markedly expanded myeloid lineage with orderly maturation; increased basophils and eosinophils; megakaryocytes may be increased
- Spleen: Massive splenomegaly (extramedullary hematopoiesis); may weigh several kilograms
- Liver: Hepatomegaly due to extramedullary hematopoiesis
5.7 Laboratory Findings
- WBC markedly elevated (50,000–200,000/μL)
- Low or absent leukocyte alkaline phosphatase (LAP) score — distinguishes CML from leukemoid reactions
- Basophilia, eosinophilia
- Philadelphia chromosome on cytogenetics; BCR::ABL1 by PCR or FISH
- Mild anemia; thrombocytosis common early
5.8 Clinical Features
- Often insidious onset
- Fatigue, weight loss, night sweats
- Symptomatic from massive splenomegaly — early satiety, left upper quadrant pain
- May be discovered incidentally on routine CBC
- Blast crisis: fever, infections, bleeding — clinically identical to acute leukemia
5.9 Treatment & Pharmacology — TKI Revolution
CML treatment was transformed by the development of imatinib, the first targeted kinase inhibitor in oncology.
| Generation | Drug | Notes |
|---|
| 1st generation | Imatinib (Gleevec) | First BCR-ABL TKI; standard of care; ~90% complete cytogenetic response |
| 2nd generation | Dasatinib, Nilotinib, Bosutinib | More potent; used for imatinib resistance/intolerance; active against many resistance mutations |
| 3rd generation | Ponatinib | Active against T315I "gatekeeper" mutation |
| STAMP inhibitor | Asciminib | Allosteric BCR-ABL inhibitor; active against T315I |
Pharmacy note: Imatinib competitively inhibits the ATP-binding site of BCR-ABL, locking the kinase in an inactive conformation. Resistance frequently arises from point mutations in the kinase domain, especially T315I, which blocks TKI binding.
Monitoring: Treatment response is monitored by quantitative RT-PCR for BCR-ABL transcripts. A major molecular response (MMR) is defined as BCR-ABL/ABL ratio ≤0.1% (IS).
5.10 Prognosis
- With modern TKI therapy: near-normal life expectancy for chronic-phase CML
- Treatment-free remission (TFR) is achievable in ~40–60% of patients who achieve deep molecular remission
- Blast crisis: median survival only 3–6 months even with treatment
6. CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) / SMALL LYMPHOCYTIC LYMPHOMA (SLL)
6.1 Definition
CLL and SLL represent the same disease at different stages. When the neoplastic cells involve the blood and bone marrow, it is called CLL; when they are confined to lymph nodes, it is called SLL. Both are composed of small, mature-appearing B lymphocytes that are functionally incompetent.
6.2 Epidemiology
- Most common leukemia in adults in the Western world
- Median age at diagnosis: 65–70 years
- Rare before age 50
- More common in males (2:1 ratio)
- Strong familial predisposition — first-degree relatives have 8-fold increased risk
6.3 Pathogenesis & Molecular Features
- Cell of origin: mature naïve or antigen-experienced B lymphocyte
- Cells are anti-apoptotic due to overexpression of BCL-2, driven by deletions of miR-15a and miR-16-1 on chromosome 13q14 (which normally suppress BCL-2)
- Frequently express CD5 (normally a T-cell marker) — a hallmark of CLL
- Driver mutations: del(13q14) — most common (favorable); del(11q) (involves ATM — unfavorable); del(17p)/TP53 (very poor prognosis); trisomy 12 (intermediate)
- Mutation status of IGHV gene is a key prognostic marker:
- Mutated IGHV: favorable prognosis (post-germinal center origin)
- Unmutated IGHV: unfavorable prognosis (pre-germinal center origin)
- ZAP-70 and CD38 expression correlate with unmutated IGHV and worse prognosis
6.4 Morphology
- Peripheral blood: Absolute lymphocytosis (>5,000 monoclonal B lymphocytes/μL); small, round lymphocytes with scant cytoplasm and clumped ("soccer ball") chromatin; smudge (basket) cells — fragile CLL cells disrupted during smear preparation
- Bone marrow: Diffuse infiltration in advanced disease
- Lymph nodes: Diffuse effacement by small lymphocytes; proliferation centers (pseudofollicles) are characteristic and contain larger, proliferating cells (prolymphocytes and paraimmunoblasts)
6.5 Immunophenotype
- CD5+, CD19+, CD20 (dim)+, CD23+, surface IgM/IgD (dim)+
- Co-expression of CD5 and CD19 is the hallmark of CLL
- Low-level surface immunoglobulin (distinguishes from mantle cell lymphoma, which is CD5+ but has bright surface Ig)
6.6 Clinical Features
- Often asymptomatic — discovered incidentally on CBC
- Progressive fatigue, lymphadenopathy, hepatosplenomegaly
- Immune dysfunction:
- Hypogammaglobulinemia → recurrent bacterial infections (most common cause of death)
- Autoimmune hemolytic anemia (warm AIHA) — Coombs-positive
- Immune thrombocytopenia (ITP)
- Richter transformation: 5–10% transform to an aggressive large B-cell lymphoma (DLBCL) — rapid clinical deterioration, poor prognosis
6.7 Staging (Rai System)
| Stage | Features | Risk |
|---|
| 0 | Lymphocytosis alone | Low |
| I | + Lymphadenopathy | Intermediate |
| II | + Splenomegaly/hepatomegaly | Intermediate |
| III | + Anemia (Hgb <11 g/dL) | High |
| IV | + Thrombocytopenia (Plt <100,000) | High |
6.8 Treatment & Pharmacology
Early-stage (Rai 0–II) CLL requires watch-and-wait — treatment does not improve survival in asymptomatic patients.
| Approach | Agents |
|---|
| BTK inhibitors (first-line for most patients) | Ibrutinib (1st gen), Acalabrutinib, Zanubrutinib (2nd gen, fewer off-target effects) |
| BCL-2 inhibitor | Venetoclax (most effective against del(17p)/TP53 disease) |
| Anti-CD20 monoclonal antibodies | Obinutuzumab, Rituximab (combined with chemo or targeted agents) |
| Older chemoimmunotherapy | FCR regimen (Fludarabine + Cyclophosphamide + Rituximab) — now largely replaced by TKIs/BCL-2 inhibitors |
| Richter transformation | Aggressive salvage chemotherapy + allogeneic SCT |
Pharmacy note: Ibrutinib irreversibly inhibits Bruton's tyrosine kinase (BTK), a key signaling molecule downstream of the B-cell receptor (BCR). BTK inhibition deprives CLL cells of survival signals and causes them to exit their lymph node niches, initially raising lymphocyte counts — a class effect that should not be mistaken for disease progression.
6.9 Prognosis
- Highly variable; median survival ranges from 2–3 years (high-risk) to >10 years (low-risk)
- Favorable: mutated IGHV, del(13q14), Rai stage 0
- Unfavorable: del(17p)/TP53, unmutated IGHV, Rai stage III–IV, Richter transformation
7. COMPARATIVE SUMMARY TABLE
| Feature | ALL | AML | CML | CLL |
|---|
| Peak age | Children (<15 yr) | Adults (>60 yr) | Adults (50–60 yr) | Elderly (>65 yr) |
| Cell lineage | Lymphoid (B or T) | Myeloid | Myeloid (HSC) | Lymphoid (B cell) |
| Key molecular marker | BCR::ABL1 (Ph+ ALL), ETV6::RUNX1, NOTCH1 | PML::RARA (APL), FLT3-ITD, NPM1 | BCR::ABL1 t(9;22) | del(13q), BCL-2 overexpression |
| Peripheral smear | Lymphoblasts | Myeloblasts + Auer rods | Full myeloid spectrum, basophilia | Smudge cells, small lymphocytes |
| Bone marrow | Lymphoblast infiltration | Myeloblast infiltration (>20%) | Hypercellular, granulocytic hyperplasia | Lymphocytic infiltration |
| Key clinical sign | Lymphadenopathy, mediastinal mass (T-ALL) | DIC (APL), gingival hyperplasia | Massive splenomegaly | Lymphadenopathy, AIHA |
| Signature drug | ATRA (APL — AML), TKIs (Ph+ ALL) | ATRA + ATO (APL), Midostaurin (FLT3+) | Imatinib / Dasatinib | Ibrutinib, Venetoclax |
| Prognosis | Excellent in children (~90%) | Intermediate (25–30%) | Excellent with TKIs | Variable (2–15 yr) |
8. TUMOR LYSIS SYNDROME — A PHARMACIST'S CONCERN
Rapid cell death during chemotherapy releases intracellular contents, leading to:
- Hyperuricemia — uric acid crystallizes in renal tubules → acute kidney injury
- Hyperkalemia → cardiac arrhythmias
- Hyperphosphatemia → hypocalcemia → tetany, seizures
Prevention and management:
- Allopurinol — xanthine oxidase inhibitor; reduces uric acid production (prophylaxis)
- Rasburicase — recombinant urate oxidase; rapidly degrades uric acid (treatment and high-risk prophylaxis); contraindicated in G6PD deficiency
- Aggressive IV hydration
- Urinary alkalinization (controversial)
9. REFERENCES
- Robbins, Cotran & Kumar. Pathologic Basis of Disease, 10th edition. Elsevier, 2023 (pp. 556–588)
- Robbins & Kumar. Basic Pathology, 11th edition. Elsevier, 2023 (pp. Chapter 13)
- Katzung, Masters & Trevor. Basic and Clinical Pharmacology, 16th edition. McGraw-Hill, 2021 (Chapter 55 — Cancer Chemotherapy)
Note to students: For the pharmacy perspective, pay particular attention to the mechanism-based treatments (ATRA in APL, TKIs in CML and Ph+ ALL, BTK inhibitors in CLL, BCL-2 inhibitors) as these represent transformative drug classes that directly exploit the molecular lesion of each leukemia subtype.