Ethiopathogenesisi of squamous intraepithelial lesion
Reading File
Finding Sources
Finding Sources
Searching PubMed
"squamous intraepithelial lesion" AND pathogenesis AND HPV
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, well-sourced material to deliver a thorough answer. Let me compose it.
Etiopathogenesis of Squamous Intraepithelial Lesion (SIL)
Source: Robbins & Kumar Basic Pathology, Chapter 17 (Female Genital System)
1. Overview and Terminology
Squamous Intraepithelial Lesion (SIL) is a spectrum of precancerous epithelial changes of the squamous epithelium, most commonly of the uterine cervix. It is classified in two parallel systems:
| Two-tier (Bethesda) | Three-tier (CIN) | Meaning |
|---|---|---|
| LSIL | CIN I | Mild dysplasia, lower 1/3 of epithelium |
| HSIL | CIN II | Moderate dysplasia, lower 2/3 |
| HSIL | CIN III | Severe dysplasia / carcinoma in situ, full thickness |
SIL peaks in incidence at ~30 years of age; invasive carcinoma peaks at ~45 years, reflecting the multi-year lag between precursor and cancer. - Robbins & Kumar Basic Pathology, p. 686
2. Etiology - The Role of HPV
Human Papillomavirus (HPV) is the single most important etiologic factor in SIL and invasive cervical carcinoma.
HPV Biology
- HPV is a DNA virus. Strains are classified as high-risk (oncogenic) or low-risk (non-oncogenic) based on genotype.
- High-risk types: HPV 16 and 18 account for ~70% of all CIN and cervical carcinoma cases.
- Low-risk types (6, 11) cause condylomas (genital warts) but almost never cause cancer.
How Infection Occurs
- Transmission is primarily through sexual contact.
- Risk factors directly tied to HPV exposure include:
- Early age at first intercourse
- Multiple sexual partners (or a partner with multiple previous partners)
- Persistent infection with high-risk HPV strains
Infection Outcomes
The following flowchart from Robbins shows the branching consequences of HPV infection:
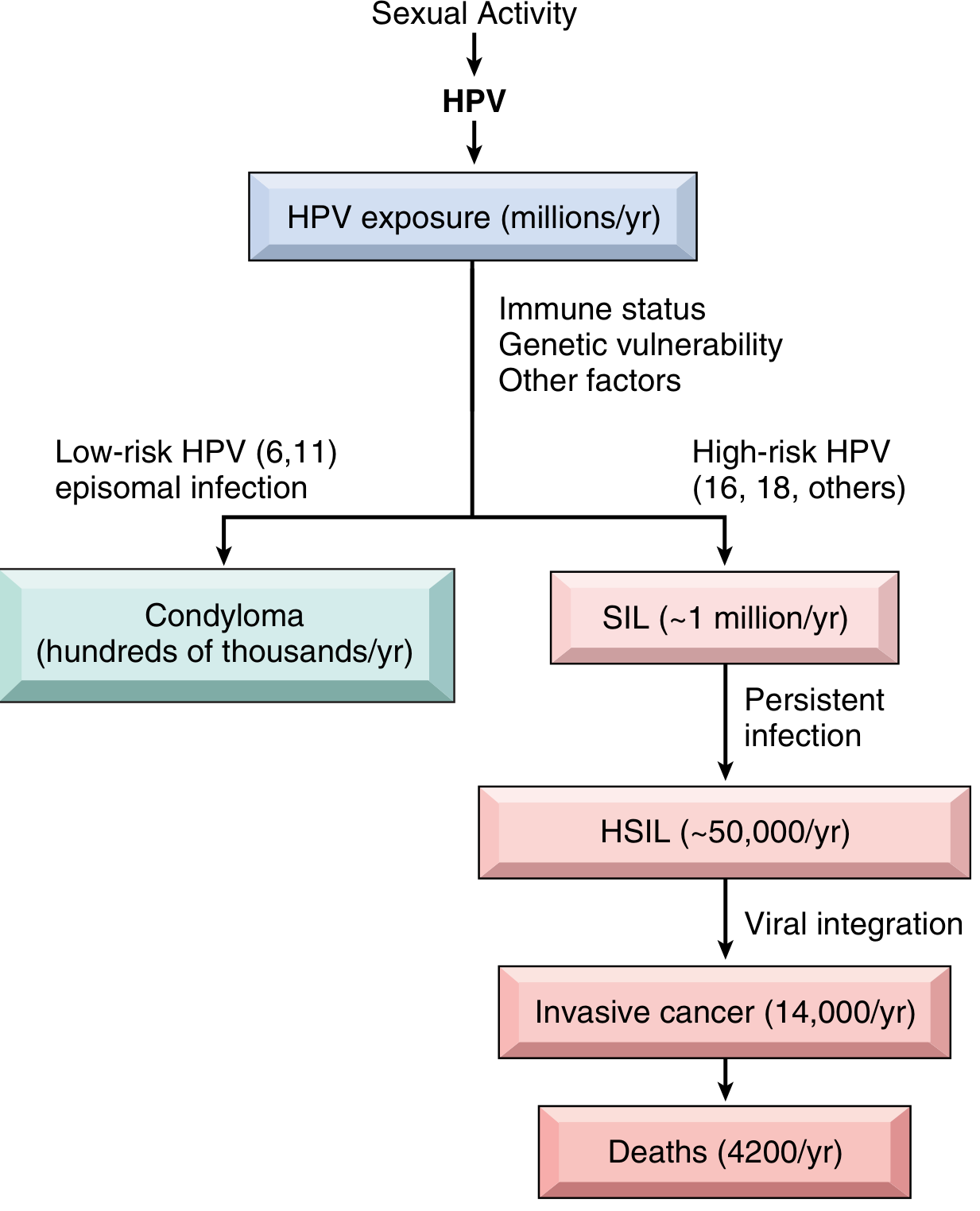
Most HPV infections are transient and cleared within months by host immunity. Only persistent infection drives progression to SIL. High-risk HPV types take longer to clear than low-risk types, increasing the window for neoplastic transformation.
3. Molecular Pathogenesis - The E6 and E7 Oncoproteins
The carcinogenic power of HPV resides entirely in two early viral proteins: E6 and E7.
Site of Action
- HPV infects immature squamous cells (at the transformation zone/squamo-columnar junction).
- Viral replication occurs in maturing squamous cells, which are normally arrested in G1.
- E6 and E7 override this G1 arrest to allow replication of the viral genome.
E6 Protein - Inactivates p53
- E6 binds to and mediates degradation of p53 (the "guardian of the genome").
- Normally p53 halts the cell cycle in response to DNA damage and promotes apoptosis.
- E6 also upregulates telomerase, conferring replicative immortality.
- Result: cells with DNA damage continue dividing instead of undergoing apoptosis.
E7 Protein - Inactivates RB
- E7 binds to the retinoblastoma protein (RB), displacing the E2F transcription factors normally sequestered by RB.
- Free E2F drives cells through the cell cycle, promoting uncontrolled proliferation.
- Result: cells bypass the G1/S checkpoint and proliferate regardless of DNA integrity.
High-risk HPV E7 has much higher affinity for RB than low-risk HPV E7. Low-risk HPV E6 fails to bind p53 at all and instead dysregulates growth via the Notch signaling pathway. This mechanistic difference explains why low-risk types do not cause cancer.
4. Viral Integration - The Key Step Toward Malignancy
Low-risk HPV types remain as free episomal DNA (extrachromosomal circles) and do not integrate into the host genome.
High-risk HPV types (16, 18) integrate into the host genome, and this integration is strongly linked to malignant progression. Integration contributes to transformation in two ways:
- Disrupts the E2 gene - E2 normally represses transcription of E6 and E7. Loss of E2 leads to increased expression of E6 and E7 oncoproteins.
- Causes genomic instability - Integration promotes accumulation of additional pro-oncogenic mutations in the host cell.
5. Co-factors and Host Factors
HPV infection alone is necessary but not sufficient for cancer. The vast majority of women infected with high-risk HPV never develop SIL or cancer. Additional co-factors influence whether infection regresses or progresses:
| Co-factor | Mechanism |
|---|---|
| Cigarette smoking | Carcinogen exposure to cervical epithelium |
| Immunocompromise (HIV, immunosuppression) | Impaired immune surveillance of HPV-infected cells |
| Genetic susceptibility | Host genetic variation in immune response |
| HPV type persistence | Higher-risk genotypes persist longer |
6. Natural History: LSIL vs. HSIL
| Lesion | Regress | Persist | Progress |
|---|---|---|---|
| LSIL (CIN I) | 60% | 30% | 10% (to HSIL) |
| HSIL (CIN II, III) | 30% | 60% | 10% (to carcinoma) |
Progression within 2-10 years. - Robbins & Kumar Basic Pathology, Table 17.1
Key biological differences:
- LSIL: High level of viral replication, mild host cell alteration, NOT considered premalignant. Koilocytic change (pathognomonic of HPV) is prominent.
- HSIL: Increased cell proliferation, arrested epithelial maturation, lower viral replication, HIGH risk of malignant transformation. Approximately 20% of HSIL develop de novo, without a detectable prior LSIL phase.
7. Histopathology of SIL
The histologic spectrum is illustrated here:

- Healthy: Orderly maturation from basal to superficial layers; abundant glycogenated cytoplasm in superficial cells.
- LSIL (CIN I): Dysplastic changes confined to the lower 1/3 of the epithelium + koilocytic change (perinuclear halos, nuclear enlargement) in superficial layers - reflecting active viral replication.
- HSIL (CIN II): Dysplastic immature cells extending to the lower 2/3 of the epithelium.
- HSIL (CIN III): Dysplastic changes involving full thickness of epithelium; marked nuclear pleomorphism, abnormal mitoses, absence of koilocytic change (viral replication is suppressed), and no surface maturation.
8. Summary of Pathogenetic Sequence
Sexual activity → HPV acquisition
↓
Infection of transformation zone (squamo-columnar junction)
↓
Transient infection → Immune clearance (most cases)
↓ (persistent high-risk HPV)
E6 destroys p53 + E7 inactivates RB
↓
Uncontrolled proliferation → LSIL (CIN I)
↓ (persistent infection + cofactors)
Viral integration → disrupts E2 → ↑E6/E7
↓
HSIL (CIN II/III) → genomic instability + mutations
↓ (10% of HSIL)
Invasive squamous cell carcinoma
Recent literature note: A 2025 systematic review (PMID 40452584) highlights that microbiome changes (vaginal dysbiosis) are associated with HPV persistence and cervical carcinogenesis - an emerging co-factor not fully covered in classic pathology texts.
- Robbins & Kumar Basic Pathology, pp. 685-688
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 4 (Oncogenic DNA Viruses)
This is a shared conversation. Sign in to Orris to start your own chat.