Retinitis pigmentosa
Reading File
search_medical_text
retinitis pigmentosa genetics pathophysiology clinical features diagnosis
search_medical_text
retinitis pigmentosa management treatment gene therapy prognosis
search_medical_image
retinitis pigmentosa fundus OCT visual field
GradeResponse
GradeResponse
GradeResponse
Retinitis Pigmentosa (RP)
Overview
Retinitis pigmentosa is a collective term for a heterogeneous group of inherited rod-cone dystrophies causing progressive photoreceptor degeneration. Despite its name, it is not an inflammatory condition — the "pigmentosa" refers to characteristic pigmentary deposits seen on fundoscopy. (Harrison's, p. 974)
Genetics & Pathophysiology
RP is genetically diverse, with >90 causative genes identified. Inheritance patterns include:
| Pattern | Proportion | Key Genes |
|---|---|---|
| Autosomal recessive | ~50–60% | ABCA4, USH2A, CNGB1 |
| Autosomal dominant | ~30–40% | RHO (rhodopsin), PRPF31, PRPH2 (peripherin) |
| X-linked | ~5–15% | RPGR, RP2 (most severe phenotype) |
| Sporadic/mitochondrial | Variable | Multiple |
Mutations in rhodopsin (the rod photopigment) and peripherin (a glycoprotein in photoreceptor outer segments) are among the most commonly identified. (Harrison's, p. 974)
Pathophysiologic sequence:
- Mutant gene → dysfunctional/misfolded protein in rod outer segments
- Rod photoreceptor apoptosis → peripheral visual loss
- Secondary cone photoreceptor degeneration (mechanism debated — oxidative stress, loss of trophic support)
- Retinal pigment epithelium (RPE) dysfunction → pigment migration into retina
Clinical Features
Symptoms (typically appear in adolescence to early adulthood):
- Nyctalopia (night blindness) — earliest and cardinal symptom; due to rod loss
- Progressive peripheral visual field loss → tunnel vision
- Photopsia (flickering lights)
- Loss of visual acuity — later finding, when foveal cones are affected
Signs on fundoscopy (classic triad):
- Bone-spicule pigmentation — irregular black deposits in mid-peripheral retina
- Arteriolar attenuation — narrowed retinal vessels
- Waxy disc pallor — optic disc appears pale/waxy
Multimodal Imaging
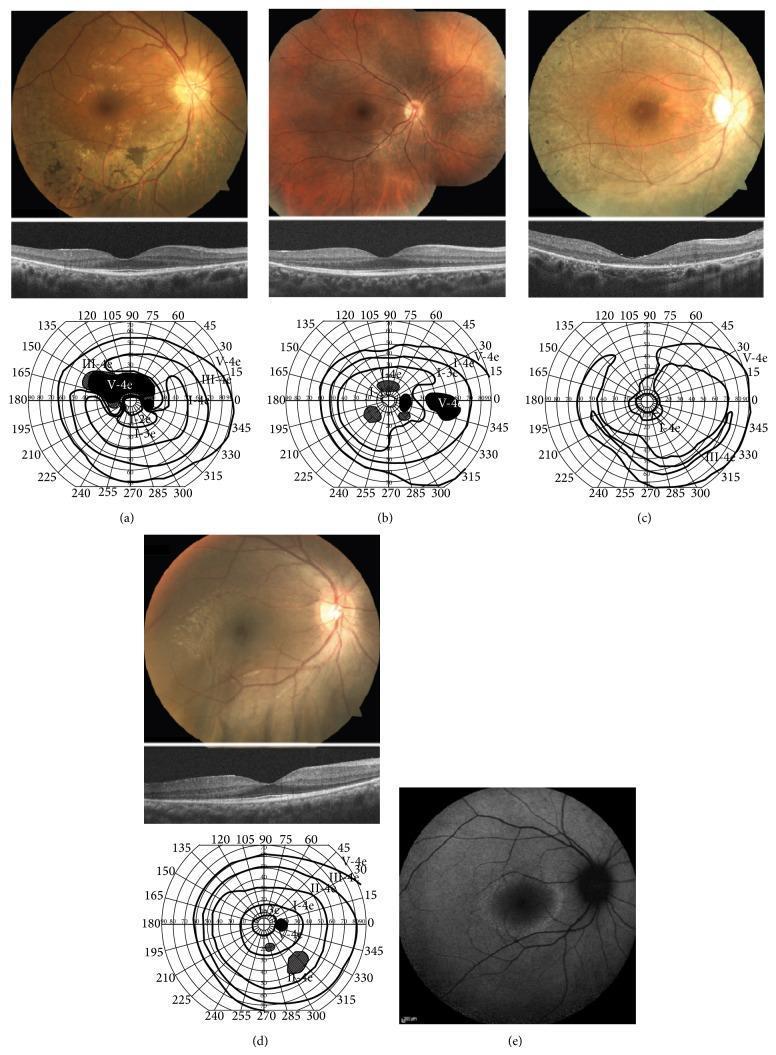
Top row: Fundus photographs showing bone-spicule pigmentary changes and arteriolar attenuation. Middle row: OCT showing marked thinning of the outer nuclear layer (ONL) with disruption/loss of the IS/OS junction, sparing only the fovea. Bottom: Goldmann perimetry demonstrating ring scotomas and peripheral constriction. Right: Fundus autofluorescence (FAF) showing a characteristic perifoveal hyperautofluorescent ring.
Diagnosis
| Test | Finding |
|---|---|
| Electroretinogram (ERG) | Severely reduced or absent rod (scotopic) responses; gold standard for diagnosis and monitoring |
| Visual field testing (Goldmann/Humphrey) | Ring scotoma progressing to tunnel vision |
| OCT | ONL thinning, IS/OS junction loss; foveal sparing until late |
| Fundus autofluorescence | Perifoveal hyperautofluorescent ring (marks zone of active degeneration) |
| Genetic testing | Identifies specific mutation; guides prognosis and eligibility for gene therapy |
Associated Syndromes
RP can be an isolated finding or part of a syndrome:
- Usher syndrome — RP + sensorineural hearing loss (most common syndromic RP; USH2A)
- Bardet-Biedl syndrome — RP + obesity + polydactyly + renal anomalies + cognitive impairment
- Kearns-Sayre syndrome — RP + external ophthalmoplegia + cardiac conduction defects (mitochondrial)
- Refsum disease — RP + peripheral neuropathy + cerebellar ataxia (phytanic acid metabolism defect)
Management
Supportive & Pharmacologic
- Vitamin A palmitate (15,000 IU/day): Modestly slows ERG deterioration; no proven benefit on visual acuity or visual fields. Use with caution (hepatotoxicity, teratogenicity). (Harrison's, p. 974)
- DHA (docosahexaenoic acid) supplementation: adjunctive, evidence limited
- UV-blocking sunglasses: Reduce phototoxic stress
- Low vision aids: Magnifiers, contrast enhancement, mobility training
- Carbonic anhydrase inhibitors (e.g., acetazolamide): For associated cystoid macular edema
Gene Therapy
- Voretigene neparvovec (Luxturna): FDA-approved (2017) for biallelic RPE65-mutation–associated retinal dystrophy. Subretinal AAV2 delivery. Represents a landmark proof-of-concept for retinal gene therapy, though RPE65 mutations account for only ~1–2% of RP cases.
- Numerous other gene therapy trials ongoing for RPGR, CNGB3, CNGA3, and other targets.
Emerging Therapies
- Retinal prostheses (e.g., Argus II / Second Sight): Epiretinal electrode array for end-stage RP; provides rudimentary light perception
- Optogenetic therapy: Introducing light-sensing proteins (channelrhodopsins) into surviving retinal cells; early clinical trials show promise
- Stem cell / RPE transplantation: Under investigation
- CRISPR gene editing: Preclinical research stage
Prognosis
- Highly variable, depending on mutation type and inheritance pattern
- X-linked RP tends to be most severe (earlier onset, faster progression)
- Most patients retain some useful central vision until the 5th–7th decade
- Legal blindness (visual acuity <20/200 or field <20°) eventually develops in the majority
- Associated cystoid macular edema and posterior subcapsular cataracts can cause additional, treatable vision loss
Key Pearls
- ERG is the gold standard — can be diagnostic even before symptoms appear
- Always screen for syndromic associations (hearing test, renal function, cardiac evaluation)
- Genetic testing is increasingly important for prognosis and treatment eligibility
- Avoid vitamin A in pregnancy; monitor liver function with supplementation
This is a shared conversation. Sign in to Orris to start your own chat.