Albinism
Reading File
Finding Sources
Searching PubMed
albinism oculocutaneous genetics management
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have excellent content from multiple authoritative textbooks. Let me compile the comprehensive answer.
Albinism
Albinism is a genetically heterogeneous group of disorders in which defects in melanin synthesis lead to partial or complete absence of pigment from the skin, hair, and/or eyes. It is not a single disease but a spectrum of conditions unified by impaired melanogenesis.
Biochemical Basis
Melanin is synthesized from tyrosine through a pathway requiring the copper-dependent enzyme tyrosinase, which catalyzes the first two rate-limiting steps. A defect in tyrosinase - or in the melanosomal proteins that support it - blocks this pathway, reducing or abolishing melanin production. The clinical result depends on which step is affected and how much residual enzyme activity remains.
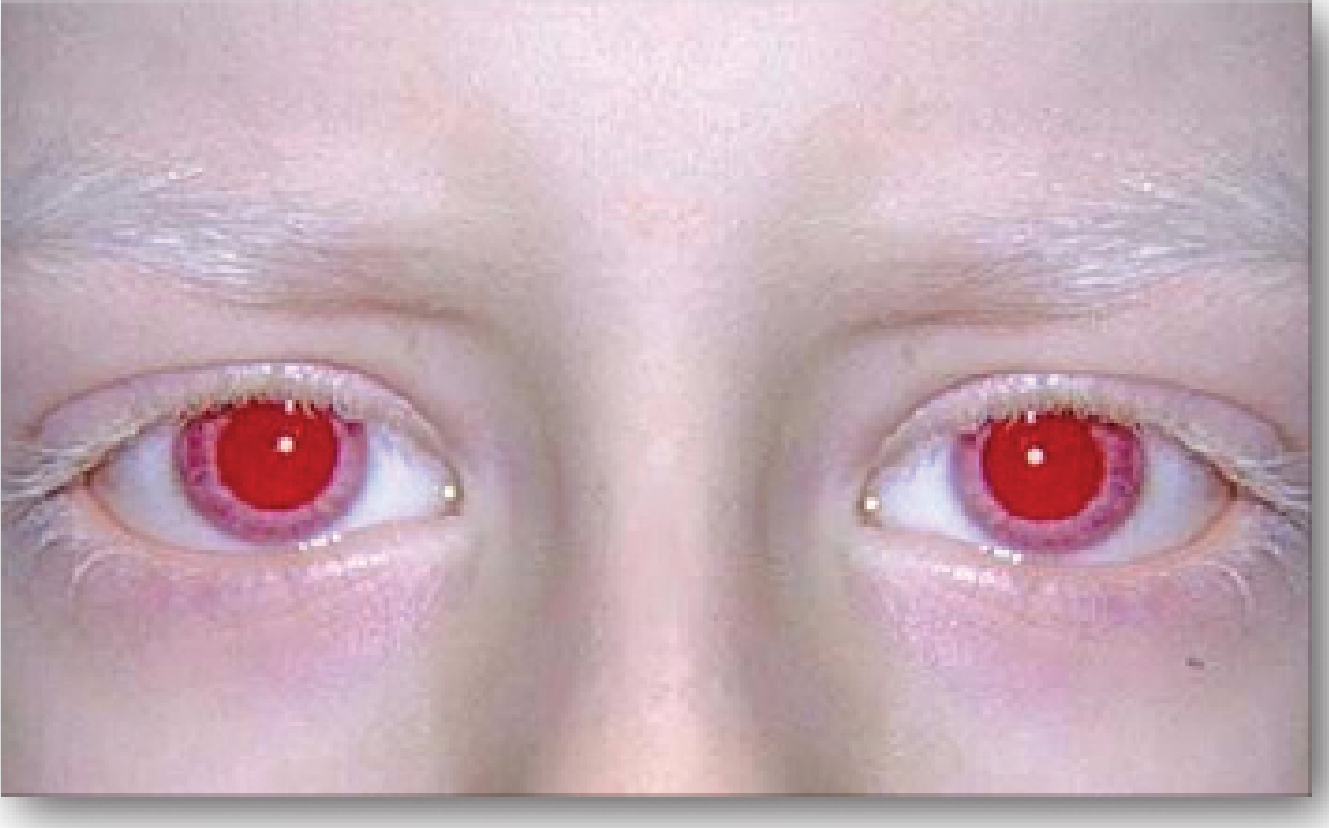
Classification
A. Oculocutaneous Albinism (OCA)
Affects skin, hair, and eyes. Inherited almost always as autosomal recessive. Seven distinct subtypes (OCA1-7) are defined by molecular genetics.
| Type | Gene | Key Features | Prevalence |
|---|---|---|---|
| OCA1A | TYR (tyrosinase) | Complete absence of tyrosinase activity; white hair/skin throughout life; pink/translucent irides; VA ~20/400 | ~1 in 40,000 (most severe) |
| OCA1B | TYR | Residual tyrosinase activity; pigment accumulates with age; can tan; hair may darken at 1-3 yrs | - |
| OCA2 | OCA2 (P-gene, chr 15q12) | Tyrosinase-positive; variable pigment from near-normal to nearly absent; most common worldwide (~50%); most common in Africa (~1 in 4,000) | ~1 in 36,000 in Europeans |
| OCA3 | TYRP1 (chr 9p23) | "Rufous/red" OCA; red hair + reddish-brown skin; mainly in Africans; visual abnormalities may be mild | Rare |
| OCA4 | MATP/SLC45A2 | Phenotypically identical to OCA2; variable hypopigmentation | Common in Japan |
| OCA5 | Chr 4q24 | Described in a Pakistani family; golden hair, nystagmus, photophobia | Single family reported |
| OCA6 | SLC24A5 | Diverse ethnicities; hair ranges from white to dark brown | Rare |
| OCA7 | C10orf11 | Leucine-rich repeat protein mutation | Rare |
OCA1 accounts for ~40% of OCA worldwide and is the most common type in Japanese and non-Hispanic Caucasians. OCA2 is the single most common form globally (~50%) and is especially prevalent in sub-Saharan Africa. - Andrews' Diseases of the Skin, p. 1009
OCA1 Subtypes in detail:
- OCA1A: Complete tyrosinase null - lifelong white hair, white skin that cannot tan, pink-to-blue irides fully translucent, VA typically 20/400
- OCA1B ("yellow mutant"): Reduced but present enzyme activity; some pigment accumulation starting age 1-3 years, tanning possible, iris may darken. A temperature-sensitive variant (OCA1-TS) produces enzyme active only below 37°C - acral areas (legs, arms, chest) develop darker hair at puberty while hair elsewhere remains white.
B. Ocular Albinism (OA)
Involvement is predominantly or exclusively ocular; skin and hair are normal (occasional hypopigmented skin macules may occur). Inherited as X-linked.
- OA1 (Nettleship-Falls syndrome) - most common
- OA2 (Forsius-Eriksson syndrome)
Affected males have hypopigmented irides and fundi. Female carriers are typically asymptomatic but may show partial iris translucency, macular stippling, and mid-peripheral areas of retinal depigmentation. - Kanski's Clinical Ophthalmology, p. 678
Ocular Features
The ocular phenotype is relatively specific across all OCA forms and includes:
- Reduced visual acuity - VA usually < 6/60 (20/200) in OCA1; caused by foveal hypoplasia (underdevelopment of the fovea, absent foveal pit, poorly formed perimacular vascular arcades)
- Nystagmus - typically pendular and horizontal; increases in bright light; tends to lessen with age
- Photophobia - due to diaphanous iris allowing excess light entry
- Iris transillumination - iris is translucent/diaphanous; "pink-eyed" appearance in severe cases
- Strabismus and absent stereopsis
- High refractive errors
- Optic chiasm misrouting - abnormal decussation of optic nerve fibers; majority of fibers from each eye cross to the contralateral hemisphere (more crossing than normal); demonstrated by abnormal (crossed) visual evoked potentials
- Fundus hypopigmentation - conspicuously visible choroidal vessels
Foveal hypoplasia and optic nerve misrouting together explain the characteristic combination of poor central acuity, nystagmus, and absent stereopsis seen even in milder forms. - Kanski's Clinical Ophthalmology, pp. 662-664; Emery's Medical Genetics, p. 1470
Syndromic Albinism (Associated Comorbidities)
Several syndromes combine albinism with potentially fatal systemic features:
Hermansky-Pudlak Syndrome (HPS)
- Autosomal recessive lysosomal storage disease
- Triad: oculocutaneous albinism + platelet dense granule deficiency (bleeding diathesis - easy bruising, excessive bleeding) + lysosomal ceroid storage
- Some subtypes develop pulmonary fibrosis and granulomatous colitis - major causes of premature mortality
- Defect in vesicular trafficking shared with melanosomes and platelet dense granules
Chediak-Higashi Syndrome (CHS)
- Autosomal recessive mutation in LYST/CHS1 gene (lysosomal regulator)
- Partial oculocutaneous albinism + giant intracellular granules in leukocytes, platelets, neurons
- Hair shows pigment clumping (large irregular melanosomes on EM) - a diagnostic clue
- Recurrent severe infections (S. aureus, streptococcus, Candida) due to impaired NK cell and CTL cytotoxicity + neutropenia + impaired leukocyte migration
- ~85% develop hemophagocytic lymphohistiocytosis (HLH) - the "accelerated phase" - during infancy/childhood; often fatal without bone marrow transplantation
- Platelet dense granule defect causes bleeding diathesis
Waardenburg Syndrome
- Autosomal dominant
- White forelock, poliosis, synophrys ("monobrow"), sensorineural deafness, limb and neurological anomalies
- Ocular features: lateral canthal displacement, iris heterochromia (segmental or complete), choroidal depigmentation
- Kanski's Clinical Ophthalmology, p. 677
Cutaneous Risks and Management
Skin Cancer Risk
All persons with OCA have significantly increased risk of basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) due to lack of photoprotective melanin. Dermoscopy is challenging given the pigment absence; a recent approach proposes:
- Biopsy any nevus with an unusual pattern or any changing nevus
- Consider reflectance confocal microscopy
Management Principles
| Domain | Intervention |
|---|---|
| Sun protection | High-SPF sunscreen, protective clothing, UV-blocking sunglasses, sun avoidance during peak hours |
| Vitamin D | Supplementation may be required due to sun avoidance |
| Ophthalmology | Corrective lenses for refractive errors, tinted lenses/sunglasses for photophobia, nystagmus may improve slightly with age; low vision aids |
| Skin surveillance | Regular dermatologic exams for BCC/SCC |
| Genetics counseling | Parents should receive genetic counseling; prenatal diagnosis possible |
| Syndromic screening | Evaluate for bleeding (HPS), infection susceptibility (CHS), hearing loss (Waardenburg) |
Genetics Summary
| Inheritance | Types |
|---|---|
| Autosomal recessive (primary) | OCA1-7; HPS; CHS |
| X-linked recessive | Ocular albinism (OA1, OA2) |
| Autosomal dominant | Waardenburg syndrome |
The most common loci are:
- Chr 11q14 - TYR gene (OCA1)
- Chr 15q12 - OCA2 gene (OCA2, also deleted in Prader-Willi/Angelman syndromes - contiguous deletion explains ~1% of Angelman/PWS patients showing albinism features)
- Chr 9p23 - TYRP1 (OCA3)
Pathophysiology Snapshot
Tyrosine
↓ [Tyrosinase - copper-dependent] ← BLOCKED IN OCA1
DOPA
↓ [Tyrosinase]
DOPAquinone
↓
Melanin
In OCA2, tyrosinase is structurally intact but the melanosomal environment is abnormal - the OCA2 protein is required for normal melanosome biogenesis, pH regulation, and processing/transport of tyrosinase itself. - Andrews' Diseases of the Skin; Lippincott Biochemistry, p. 769
Sources:
- Andrews' Diseases of the Skin, pp. 1008-1010
- Kanski's Clinical Ophthalmology (10th ed.), pp. 662-664, 677-678
- Lippincott's Illustrated Reviews: Biochemistry (8th ed.), p. 769
- Emery's Elements of Medical Genetics and Genomics, p. 1470
Give flashcard of it
Generating Image

Here's your Albinism flashcard covering all the high-yield points:
- Biochemistry - tyrosinase defect blocking melanin synthesis
- OCA types 1-7 with genes, chromosomes, and key features
- Ocular features - foveal hypoplasia, nystagmus, chiasm misrouting
- Syndromic associations - Hermansky-Pudlak, Chediak-Higashi, Waardenburg
- Inheritance patterns and management principles
This is a shared conversation. Sign in to Orris to start your own chat.