Explain Heart in full description
The Heart - A Full Description
Overview
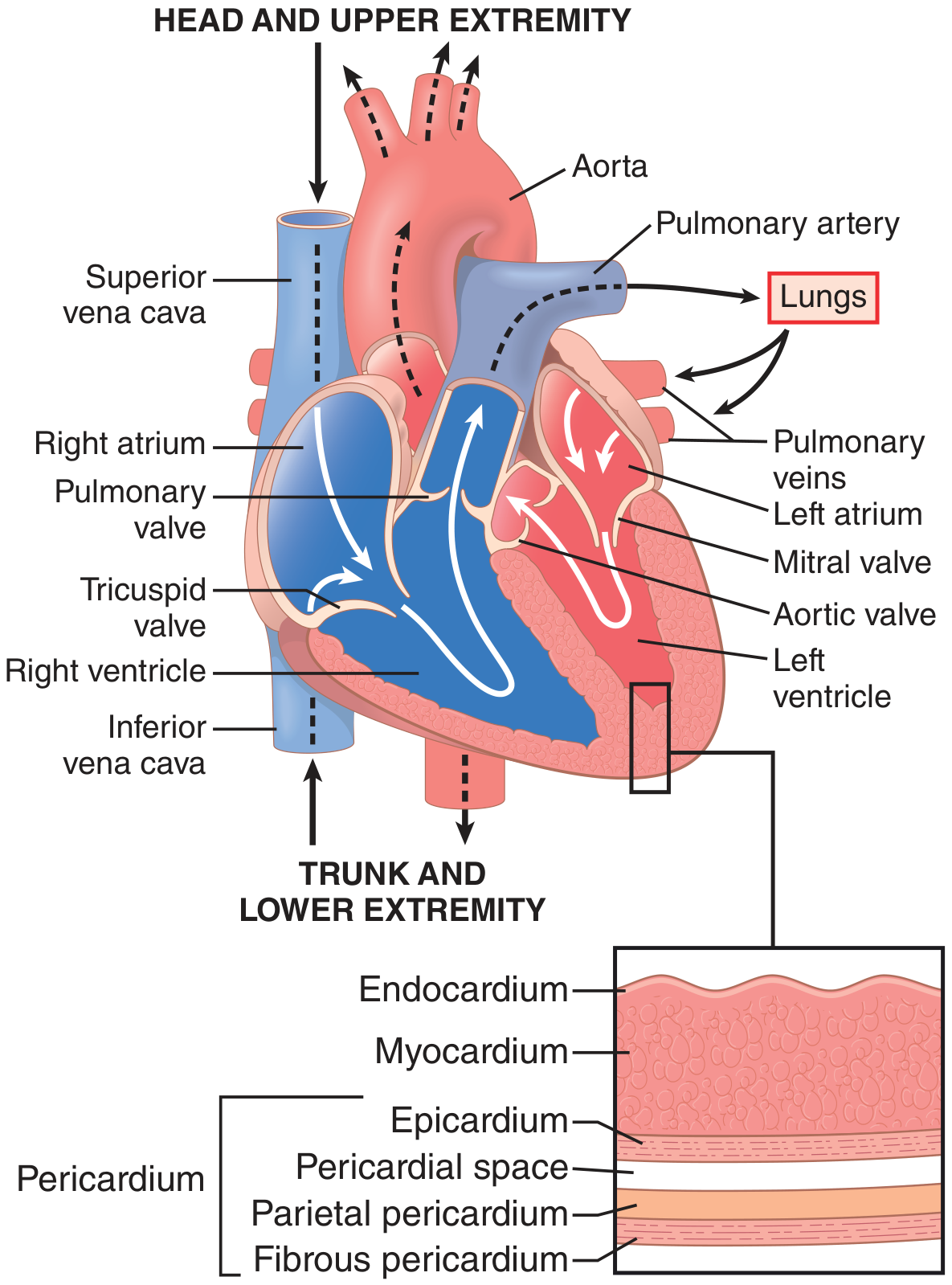
1. Location and External Anatomy
- An apex pointing inferolaterally to the left (approximately the 5th intercostal space, midclavicular line)
- A base (posterior surface) facing superiorly and posteriorly, formed mainly by the left atrium
- Four surfaces: sternocostal (anterior), diaphragmatic (inferior), left pulmonary, and right pulmonary
- Fibrous pericardium: tough outer layer, anchors the heart to surrounding structures
- Serous pericardium: inner double layer - the parietal layer lines the fibrous pericardium, and the visceral layer (epicardium) is adherent to the heart surface. The potential space between them, the pericardial cavity, normally contains 15-50 mL of serous fluid that reduces friction.
2. Wall Layers
| Layer | Description |
|---|---|
| Endocardium | Inner smooth lining of endothelial cells; lines chambers and covers valves |
| Myocardium | Thick middle layer of cardiac muscle; responsible for contraction |
| Epicardium | Outer layer (visceral pericardium); contains coronary vessels and fat |
3. Chambers
Right Atrium
Right Ventricle
Left Atrium
Left Ventricle
The left ventricle is organized into complex muscle fiber layers. The subepicardial (outer) fibers spiral in a left-handed helix and the subendocardial (inner) fibers spiral in a right-handed (opposite) helix, creating a double helix. This produces a wringing/twisting motion during systole - the apex rotates counterclockwise and the base rotates clockwise (viewed from apex). This torsion aids both ejection and rapid filling during diastole.
- Guyton & Hall Textbook of Medical Physiology
4. Valves
Atrioventricular (AV) Valves
- Tricuspid valve (right AV valve): 3 leaflets; separates right atrium from right ventricle
- Mitral (bicuspid) valve (left AV valve): 2 leaflets; separates left atrium from left ventricle
Semilunar Valves
- Pulmonary valve: 3 cusps; at the outflow of the right ventricle into the pulmonary trunk
- Aortic valve: 3 cusps; at the outflow of the left ventricle into the aorta
5. Cardiac Muscle (Myocardium) - Histology & Cell Biology
Key Features of Cardiomyocytes
- Striated: contain parallel actin (thin) and myosin (thick) filaments, organized into sarcomeres with Z lines - structurally similar to skeletal muscle
- Branching, interconnected: fibers branch and rejoin in a latticework pattern
- Intercalated discs: dark bands crossing the fibers at junctions between cells; contain gap junctions (connexins) that allow rapid ion diffusion between cells
- Functional syncytium: because ions flow freely through gap junctions, an electrical impulse spreads rapidly across all interconnected cells. The heart acts as two syncytia - atrial and ventricular - separated by the fibrous AV ring (impulses can only cross through the AV bundle)
- Single or binucleate nuclei, centrally placed (unlike peripheral nuclei in skeletal muscle)
- Rich in mitochondria: ~25-35% of cell volume, reflecting near-constant aerobic demand
Action Potential
- Fast sodium channels (phase 0 depolarization - same as skeletal muscle)
- Slow calcium channels (L-type, LTCC) that open and stay open during the plateau - calcium influx prolongs the action potential and enables contraction
Excitation-Contraction Coupling
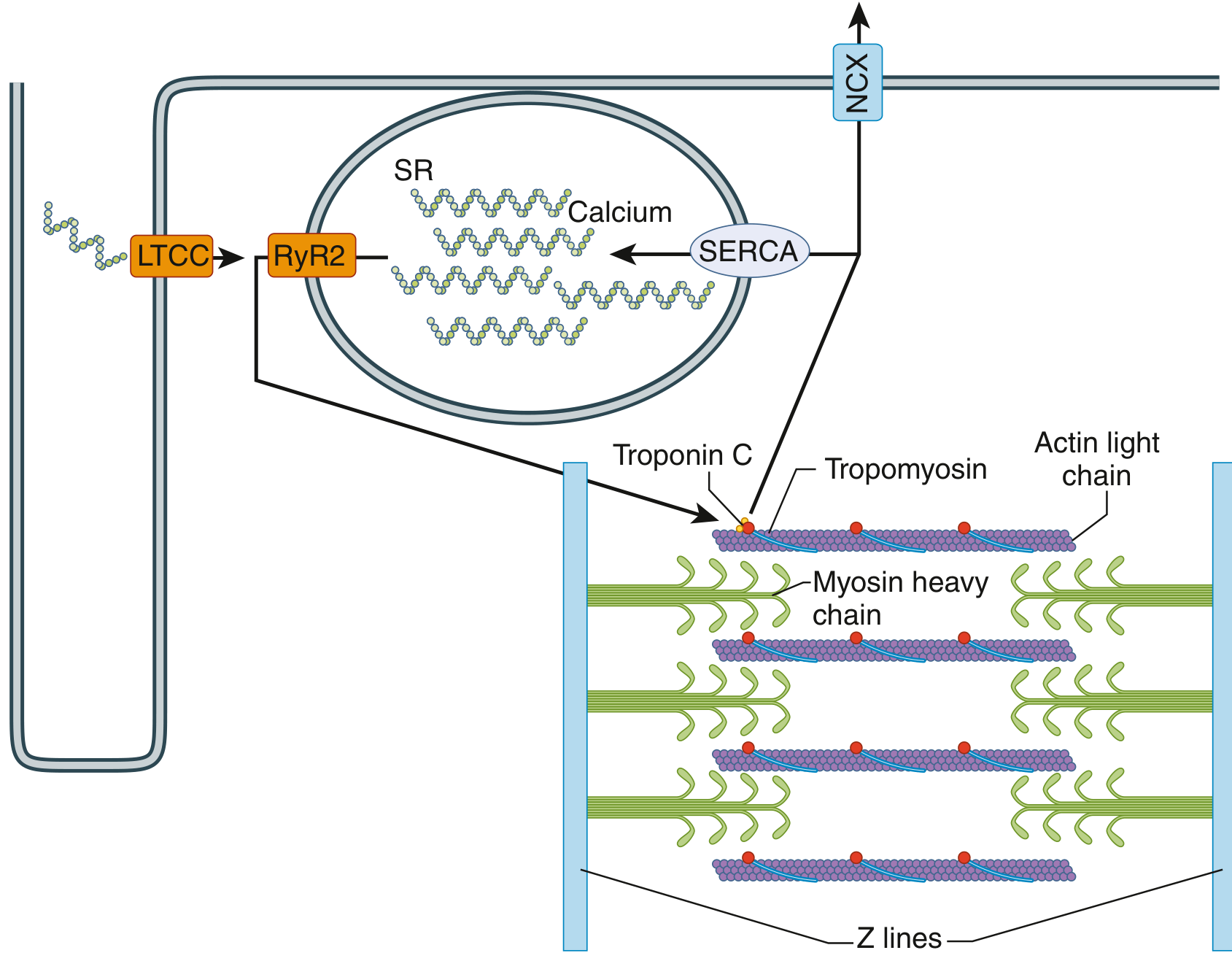
- Action potential depolarizes the cell membrane and travels into the T-tubules (transverse tubules, 5× the diameter of those in skeletal muscle)
- Depolarization opens L-type calcium channels (LTCC) in the T-tubule membrane
- Ca²⁺ entry triggers massive Ca²⁺ release from the sarcoplasmic reticulum via ryanodine receptor 2 (RyR2) - a process called "calcium-induced calcium release"
- Cytosolic Ca²⁺ binds troponin C, causing a conformational change in tropomyosin that exposes actin binding sites
- Myosin heads bind actin, forming cross-bridges, and pull actin filaments inward (sliding filament mechanism) - producing contraction
- Relaxation: SERCA (sarcoplasmic reticulum Ca²⁺-ATPase) pumps Ca²⁺ back into the SR; the sodium-calcium exchanger (NCX) pumps Ca²⁺ out of the cell
Without calcium from the T-tubules, cardiac contraction would be severely reduced, because the sarcoplasmic reticulum of cardiac muscle is less well developed than in skeletal muscle. This is why extracellular calcium concentration directly affects cardiac contractile strength - unlike skeletal muscle.
- Guyton & Hall Textbook of Medical Physiology
6. Conduction System
Sinoatrial (SA) Node - The Pacemaker
- Located at the junction of the SVC and the right atrium (superior end of the crista terminalis)
- Spontaneously depolarizes at 60-100 beats/min (intrinsic rate), setting the heart's rhythm
- Excitation signals spread across both atria, causing atrial contraction
Atrioventricular (AV) Node
- Located near the opening of the coronary sinus, close to the septal cusp of the tricuspid valve, within the AV septum
- Receives the impulse from the atria and introduces a critical delay (~0.1 s) allowing atrial contraction to finish and ventricles to fill before ventricular contraction begins
- Acts as a "gatekeeper" - the only normal electrical bridge between atria and ventricles (the fibrous AV ring otherwise insulates them)
- Intrinsic rate: 40-60 beats/min (escape rhythm if SA node fails)
Bundle of His (AV Bundle)
- A direct continuation of the AV node; runs along the lower border of the membranous interventricular septum
- Divides into right and left bundle branches
- Right bundle branch: travels down the right side of the septum, enters the septomarginal trabecula, reaches the base of the anterior papillary muscle, then spreads into Purkinje fibers of the right ventricle
- Left bundle branch: passes to the left side of the muscular septum, descends toward the apex, gives off branches to the left ventricle
Purkinje Fibers (Subendocardial Plexus)
- Final network of large, fast-conducting specialized cells spreading throughout both ventricular walls
- Conduct impulses at 2-4 m/s (much faster than regular myocardium)
- Ensure the wave of excitation and contraction moves from the papillary muscles and ventricular apex upward toward the arterial outflow tracts - this bottom-to-top sequence efficiently ejects blood
- Intrinsic rate: 20-40 beats/min (terminal escape rhythm)
The unique distribution pattern of the cardiac conduction system establishes a unidirectional pathway of excitation/contraction. Large branches are insulated from surrounding myocardium by connective tissue to prevent inappropriate stimulation.
- Gray's Anatomy for Students
7. The Cardiac Cycle
| Phase | Events | ECG | Valves | Heart Sounds |
|---|---|---|---|---|
| A - Atrial Systole | Atria contract; final ventricular filling | P wave | - | S4 (if present) |
| B - Isovolumetric Ventricular Contraction | Ventricles contract; pressure rises; all valves closed; volume unchanged | QRS complex | Mitral closes | S1 (lub) |
| C - Rapid Ventricular Ejection | Ventricular pressure exceeds aortic; blood ejected rapidly; ventricular volume falls | ST segment | Aortic opens | - |
| D - Reduced Ventricular Ejection | Slower ejection; ventricular volume reaches minimum; aortic pressure begins to fall | T wave | - | - |
| E - Isovolumetric Ventricular Relaxation | Ventricles relax; pressure falls; all valves closed; volume unchanged | - | Aortic closes | S2 (dub) |
| F - Rapid Ventricular Filling | AV valves open; ventricles fill passively; volume rises rapidly | - | Mitral opens | S3 (physiologic in young) |
| G - Reduced Ventricular Filling (Diastasis) | Slow passive filling; heart at lowest volume | - | - | - |
Heart Sounds
- S1 ("lub"): closure of mitral and tricuspid valves at onset of systole
- S2 ("dub"): closure of aortic and pulmonary valves at onset of diastole
- S3: rapid ventricular filling (normal in children; abnormal in heart failure)
- S4: atrial contraction into a stiff ventricle (always abnormal)
8. Coronary Circulation
Left Coronary Artery (LCA)
- Short main stem (left main coronary artery)
- Divides into:
- Left anterior descending (LAD) / anterior interventricular artery: runs in the anterior interventricular groove; supplies the anterior LV, anterior interventricular septum, and apex - often called the "widow maker"
- Left circumflex artery: runs in the left atrioventricular groove; supplies the lateral and posterior LV and left atrium
Right Coronary Artery (RCA)
- Runs in the right AV groove
- Gives off the right marginal artery and usually the posterior interventricular (descending) artery
- Supplies the right atrium, right ventricle, SA node (in ~60% of people), AV node (in ~80% of people), and inferior LV
Coronary Flow Regulation
- Most LV coronary flow occurs during diastole (systolic LV wall compression occludes intramural vessels); RV coronary flow continues in both systole and diastole due to lower pressures
- Coronary flow can increase up to 6-fold above resting levels during exercise, mediated by local vasodilators: nitric oxide, adenosine, bradykinins, prostaglandins, and CO₂
- The LV extracts ~70-80% of delivered oxygen at rest - near-maximal - so increased oxygen demand requires increased flow, not increased extraction
Venous Drainage
- Most venous blood drains into the coronary sinus (in the posterior AV groove), which empties into the right atrium
- Anterior cardiac veins drain directly into the right atrium
- Thebesian veins drain directly into all chambers
9. Determinants of Cardiac Performance
| Factor | Definition | Influencing Conditions |
|---|---|---|
| Heart Rate (HR) | Beats per minute | Autonomic tone, hormones, temperature |
| Preload | Volume of blood in ventricle before contraction; reflected by end-diastolic volume (EDV) | Venous return, blood volume |
| Afterload | Resistance the ventricle must overcome to eject; approximated by aortic/pulmonary pressure | Systemic vascular resistance, aortic stenosis |
| Contractility (Inotropy) | Intrinsic strength of contraction independent of loading conditions | Sympathetic stimulation, catecholamines, calcium |
10. Autonomic Innervation
- Sympathetic (thoracic ganglia T1-T4): releases norepinephrine onto β1-adrenergic receptors → increases HR (chronotropy), contractility (inotropy), and conduction velocity (dromotropy)
- Parasympathetic (vagus nerve, CN X): releases acetylcholine onto muscarinic (M2) receptors → decreases HR and slows AV conduction
11. Blood Flow Through the Heart
12. Key Clinical Correlates
- Myocardial infarction: occlusion of a coronary artery (most commonly the LAD) causes ischemia and cell death. Visceral afferent pain fibers follow sympathetic nerves (T1-T4), producing referred pain to the chest, left arm, jaw, or epigastrium
- Heart failure: inability of the heart to pump sufficient blood at normal filling pressures; results from impaired contractility (systolic failure) or impaired relaxation/filling (diastolic failure)
- Wolff-Parkinson-White syndrome: congenital accessory pathway bypasses the AV node, allowing abnormal impulses to reach the ventricles and cause potentially fatal tachyarrhythmias
- Pericardial effusion/tamponade: excess fluid in the pericardial space compresses the heart and impairs filling
- Coronary artery disease: atherosclerotic plaque narrows coronary arteries, reducing myocardial oxygen supply; can cause angina (reversible) or MI (irreversible)
- Guyton & Hall Textbook of Medical Physiology - Cardiac Muscle Physiology, Ch. 9
- Gray's Anatomy for Students - Cardiac Conduction System
- Goldman-Cecil Medicine - Anatomy of the Heart, Ch. 41
- Costanzo Physiology, 7th Ed. - The Cardiac Cycle, Ch. 4
- Barash Clinical Anesthesia, 9th Ed. - Cardiac Anatomy and Physiology, Ch. 12
Pathophysiology on Heart Failure
"heart failure"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Pathophysiology of Heart Failure
Definition
- Forward failure (low output): fatigue, dizziness, muscle weakness, impaired organ perfusion
- Backward failure (congestion): elevated filling pressures causing pulmonary or peripheral edema
Normal cardiac output is ~5 L/min. Systolic dysfunction (HFrEF) has EF <45% (normal >60%). Diastolic dysfunction (HFpEF) has reduced filling with a relatively preserved EF.
- Katzung's Basic & Clinical Pharmacology, 16th Ed.
The Multidimensional Nature of Heart Failure
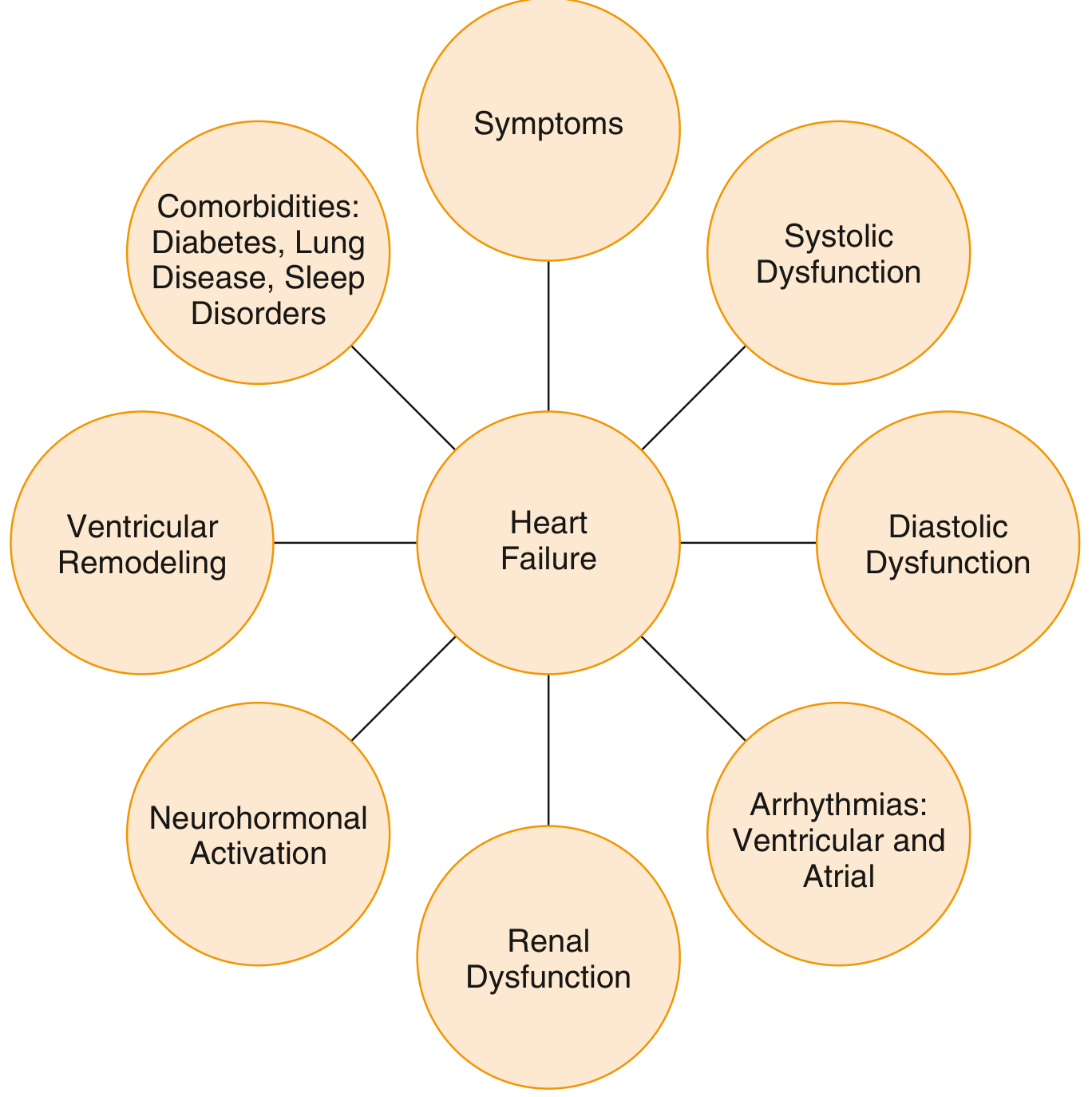
1. Common Causes (Initiating Insults)
| Category | Examples |
|---|---|
| Pressure overload | Systemic hypertension, aortic stenosis, pulmonary hypertension |
| Volume overload | Valvular regurgitation (mitral/aortic), septal defects, AV fistulas |
| Primary myocardial loss | Myocardial infarction, ischemic heart disease (IHD) |
| Cardiomyopathies | Dilated, hypertrophic, restrictive, toxic (alcohol, anthracyclines) |
| Tachyarrhythmia-mediated | Persistent atrial fibrillation with rapid ventricular response |
| Genetic/infiltrative | Familial cardiomyopathies (sarcomere mutations), amyloidosis, hemochromatosis |
| High-output states | Severe anemia, hyperthyroidism, AV shunts, beriberi |
2. Cardiac Hypertrophy - The First Adaptive Response
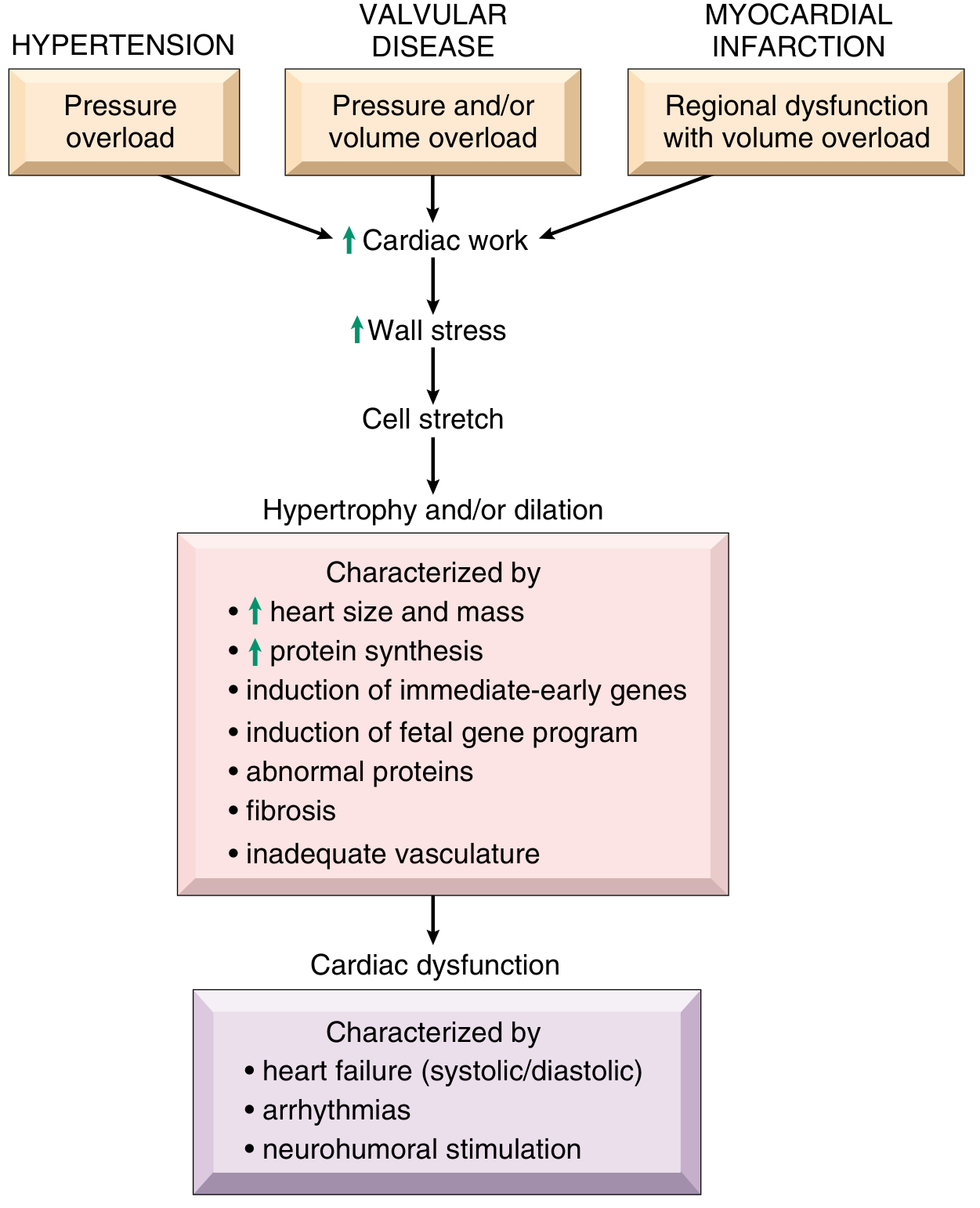
Concentric Hypertrophy (Pressure Overload)
- New sarcomeres assemble in parallel with existing ones
- Cross-sectional area of myocytes increases
- Wall thickness increases without chamber dilation
- Typical of: hypertension, aortic stenosis
- LV wall thickness can increase 2-3x normal; heart weight can be 2-4x normal
Eccentric Hypertrophy (Volume Overload)
- New sarcomeres assemble in series with existing ones
- Chamber dilates (elongation of myocytes)
- Wall thickness may be normal, increased, or decreased
- Typical of: mitral regurgitation, aortic regurgitation, dilated cardiomyopathy post-failure
Why Hypertrophy Eventually Fails
- Inadequate capillary density: Myocyte mass increases, but capillary proliferation does not keep pace → relative ischemia, especially subendocardially
- Increased metabolic demand: Greater mass, elevated heart rate, and elevated contractility all increase O₂ consumption
- Interstitial fibrosis: Mechanical stress drives myocardial fibroblasts to increase extracellular matrix synthesis → stiff, non-compliant ventricle → diastolic dysfunction
- Re-expression of fetal gene programs: Hypertrophied myocytes downregulate adult isoforms and re-express fetal forms of myosin, natriuretic peptides, and collagen - these fetal proteins are less efficient
- Immediate-early gene activation: FOS, JUN, MYC, EGR1 alter protein expression and metabolism
- Accelerated cardiomyocyte apoptosis: Neurohormones, adrenergic activation, inflammatory mediators, and toxins all accelerate cell death; loss of contractile mass worsens pump function
Cardiac hypertrophy is associated with heightened metabolic demands and inadequate capillary density, making the hypertrophied heart vulnerable to ischemia-related decompensation.
- Robbins, Cotran & Kumar Pathologic Basis of Disease
3. Neurohormonal Activation - The Vicious Cycle
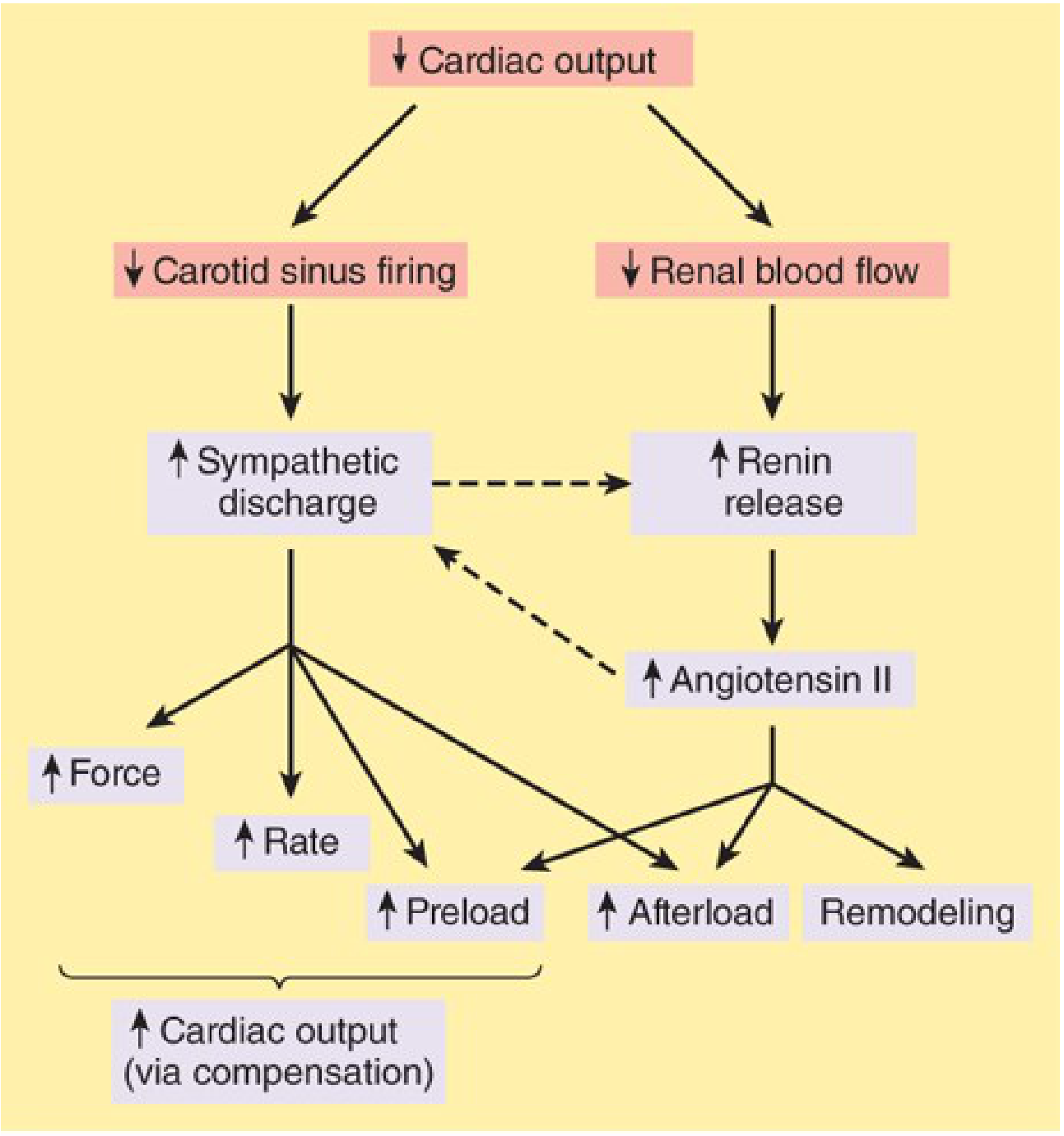
A. Sympathetic Nervous System (SNS) Activation
- Tachycardia (increased HR)
- Increased myocardial contractility
- Venoconstriction → increased venous return (preload)
- Sustained elevated norepinephrine is directly cardiotoxic
- β1-receptor downregulation and uncoupling → blunted inotropic response
- β2 receptor coupling shifts to the IP3-DAG cascade
- Excessive β activation causes calcium leak from the SR via RyR channels → arrhythmias and diastolic stiffening
- SERCA2a function impaired → impaired calcium reuptake → diastolic dysfunction
- Activates caspases → accelerated apoptosis
- SNS and RAAS are co-regulated and amplify each other
B. Renin-Angiotensin-Aldosterone System (RAAS) Activation
| Effect | Result |
|---|---|
| Angiotensin II | Potent vasoconstriction → ↑ afterload |
| Aldosterone | Sodium and water retention → ↑ preload and congestion |
| Angiotensin II | Promotes myocardial and vascular fibrosis (remodeling) |
| Angiotensin II | Stimulates further norepinephrine release from sympathetic nerve endings |
| Combined | Self-amplifying loop accelerating dysfunction |
C. Other Neurohormones
- Arginine vasopressin (AVP/ADH): released in HF; causes vasoconstriction via V1 receptors; causes water retention via V2 receptors → hyponatremia and volume overload
- Endothelin-1: potent, prolonged vasoconstrictor released by vascular endothelium; reduces glomerular filtration; causes pulmonary arteriolar constriction
- Natriuretic peptides (BNP, ANP): released by ventricular myocytes under wall stress as a counter-regulatory response; cause vasodilation, natriuresis, and diuresis. However, this system becomes overwhelmed in advanced HF. Serum BNP/NT-proBNP levels are used clinically as markers of HF severity and prognosis
- Proinflammatory cytokines (TNF-α, IL-1β, IL-6): elevated in HF; contribute to myocyte apoptosis, hypertrophy, and matrix remodeling
4. Ventricular Remodeling
- Chamber dilation (progressive with neurohormonal activation)
- Abnormal myocardial cell phenotype (biochemical characteristics of fetal myocytes)
- Proliferation of connective tissue → interstitial fibrosis
- Change in ventricular geometry: progressive sphericalization (from elliptical to spherical shape) reduces mechanical efficiency
- Mitral annular dilation: as LV dilates, the mitral annulus distorts → functional mitral regurgitation → further volume overload → further dilation (another vicious cycle)
- Progressive myocyte loss via apoptosis and necrosis → surviving myocytes bear greater wall stress
Remodeling includes proliferation of connective tissue cells and abnormal myocytes with biochemical characteristics of fetal myocytes. Ultimately, myocytes die at an accelerated rate via apoptosis, and remaining myocytes are subject to even greater stress.
- Katzung's Basic & Clinical Pharmacology, 16th Ed.
5. Cellular and Molecular Mechanisms of Contractile Failure
Calcium Cycling Abnormalities
- SERCA2a downregulation/dysfunction: impaired Ca²⁺ reuptake into the SR → elevated diastolic [Ca²⁺] → incomplete relaxation → diastolic dysfunction
- RyR2 hyperphosphorylation: causes diastolic SR calcium leak → depletes SR calcium stores → reduced systolic Ca²⁺ transient → impaired contraction
- Net result: reduced systolic [Ca²⁺] peak + elevated diastolic [Ca²⁺] → weakened contraction AND impaired relaxation
Myofilament Dysfunction
- Reduced SR Ca²⁺ content alters interaction of cardiac myosin and actin
- Fetal isoforms of myosin heavy chain (β-MHC) replace adult isoforms (α-MHC) → lower ATPase activity and slower, less efficient cross-bridge cycling
Energetic Deficiency
- Mitochondrial dysfunction impairs ATP production
- Failing heart shifts metabolism from fatty acid oxidation (normal predominant fuel) to glycolysis → less efficient energy production per oxygen molecule
- Energy depletion impairs SERCA, Na⁺/K⁺-ATPase, and actomyosin ATPase function
Ion Channel Remodeling
- Potassium channel downregulation → prolonged action potential duration → arrhythmogenic substrate
- This is a primary cause of sudden cardiac death in HF patients
6. Types of Heart Failure
HFrEF (Heart Failure with Reduced Ejection Fraction)
- EF <40%; also called "systolic heart failure"
- Dominant feature: impaired contractility (systolic dysfunction)
- Pump cannot generate adequate stroke volume
- Common causes: MI, dilated cardiomyopathy, myocarditis
- Frank-Starling curve depressed and shifted rightward
HFpEF (Heart Failure with Preserved Ejection Fraction)
- EF ≥50%; also called "diastolic heart failure"
- Dominant feature: impaired ventricular relaxation and/or increased stiffness
- The ventricle cannot fill adequately at normal filling pressures
- Mechanism: LV hypertrophy + interstitial fibrosis → ↑ chamber stiffness; impaired Ca²⁺ cycling → slow relaxation
- Common causes: hypertension (most common), aging, diabetes, obesity
- Resting hemodynamics may be near-normal, but exertion or tachycardia causes dramatic rise in filling pressures → dyspnea
- Atrial fibrillation particularly harmful: loss of atrial kick into a noncompliant ventricle markedly impairs filling
- Diuresis must be cautious: narrow window between fluid overload and underfilling
HFmrEF (Mildly Reduced EF)
- EF 41-49%; intermediate phenotype with features of both above
7. Left-Sided vs. Right-Sided Heart Failure
Left-Sided Heart Failure
- Elevated LV filling pressure → elevated left atrial pressure → elevated pulmonary capillary pressure → pulmonary congestion and edema
- Lungs: heavy, wet; perivascular and interstitial edema → alveolar edema
- Heart failure cells (hemosiderin-laden macrophages): sign of previous pulmonary edema - macrophages phagocytose RBCs and plasma proteins leaked into alveoli
- Pleural effusions from elevated pleural capillary/lymphatic pressure
- Exertional dyspnea (earliest)
- Orthopnea (dyspnea when supine - fluid redistributes from legs to lungs; relieved by sitting up)
- Paroxysmal nocturnal dyspnea (PND) (waking at night, severe breathlessness)
- Dyspnea at rest (advanced)
- Cardiac wheeze / fine crackles at lung bases
- Reduced renal perfusion → RAAS activation → salt/water retention → worsens congestion
- Severe: azotemia (prerenal), cerebral hypoperfusion (confusion, encephalopathy)
- Secondary LV dilation → functional mitral regurgitation → atrial dilation → atrial fibrillation
Right-Sided Heart Failure
- Elevated right-sided pressures → systemic venous congestion
| Organ | Effect |
|---|---|
| Subcutaneous tissue | Dependent/pedal edema, pretibial edema, sacral edema, anasarca |
| Liver | "Nutmeg liver" - centrilobular congestion (red-brown mottling); cardiac cirrhosis with chronic congestion |
| Spleen | Congestive splenomegaly |
| GI tract | Gut congestion → malabsorption, "cardiac cachexia" |
| Kidneys | Venous congestion + reduced arterial flow → azotemia, sodium retention |
| Jugular veins | Elevated JVP, hepatojugular reflux |
8. Cardiorenal Syndrome
- Reduced cardiac output → ↓ renal perfusion pressure
- Elevated venous pressure → ↑ renal venous pressure → impaired renal filtration
- Both mechanisms activate RAAS and SNS → sodium/water retention → worsening congestion
- Worsening renal function limits use of ACE inhibitors and ARBs that would otherwise slow HF progression
- Serum creatinine rise during HF treatment can represent a vicious cycle or treatment-related hemodynamic adjustment
9. Frank-Starling Mechanism in Heart Failure
- The ventricular function curve is depressed - the same preload generates less stroke volume
- Beyond ~15 mmHg filling pressure, there is a plateau
- Preloads >20-25 mmHg result in pulmonary congestion without further improvement in output
- Neurohormonal activation increases preload (sodium/water retention) in an attempt to compensate via the Starling mechanism, but this backfires by causing congestion and further increasing wall stress
10. Summary: The Self-Perpetuating Downward Spiral
Initial insult (MI, hypertension, valve disease)
↓
↓ Cardiac output
↓
Baroreceptor activation → SNS + RAAS stimulation
↓
↑ HR, ↑ contractility, ↑ vasoconstriction (afterload ↑), ↑ sodium/water retention (preload ↑)
↓
Short-term: maintained CO (compensation)
↓
Prolonged: β1-downregulation, Ca²⁺ cycling impairment, RAAS-mediated fibrosis,
myocyte apoptosis, ventricular remodeling, mitral regurgitation
↓
Further ↓ EF → more neurohormonal activation → progressive remodeling
↓
End-stage: severe biventricular failure, arrhythmias, renal failure, death
11. Biomarkers of HF Pathophysiology
| Biomarker | Source | Significance |
|---|---|---|
| BNP / NT-proBNP | Ventricular myocytes (wall stress) | Elevated: confirms HF, tracks severity and prognosis |
| Troponin I/T | Cardiomyocyte necrosis | Elevated in acute decompensation and HFrEF |
| Norepinephrine | Sympathetic activation | Elevated levels associated with worse prognosis |
| Aldosterone | RAAS activation | Drives sodium retention and myocardial fibrosis |
| Endothelin-1 | Vascular endothelium | Vasoconstriction, pulmonary hypertension |
| Creatinine/GFR | Cardiorenal syndrome | Worsening = adverse prognostic marker |
- Robbins, Cotran & Kumar - Pathologic Basis of Disease - Pathophysiology and Progression to Heart Failure, Ch. 12
- Goldman-Cecil Medicine - Pathobiologic Mechanisms of Heart Failure, Ch. 45
- Katzung's Basic & Clinical Pharmacology, 16th Ed. - Pathophysiology of Heart Failure, Ch. 13
- Goodman & Gilman's Pharmacological Basis of Therapeutics - Pathophysiology of Heart Failure
- Barash Clinical Anesthesia, 9th Ed. - Pathophysiology of Heart Failure, Ch. 12
Connective tissue It's types, Function, Cells, Fiber, ECM, and It's location
Connective Tissue - Full Description
Overview and Embryonic Origin
The variety of connective tissue types in the body reflects differences in composition and amount of the cells, fibers, and ground substance, which together are responsible for the remarkable structural, functional, and pathologic diversity of connective tissue.
- Junqueira's Basic Histology, 17th Ed.
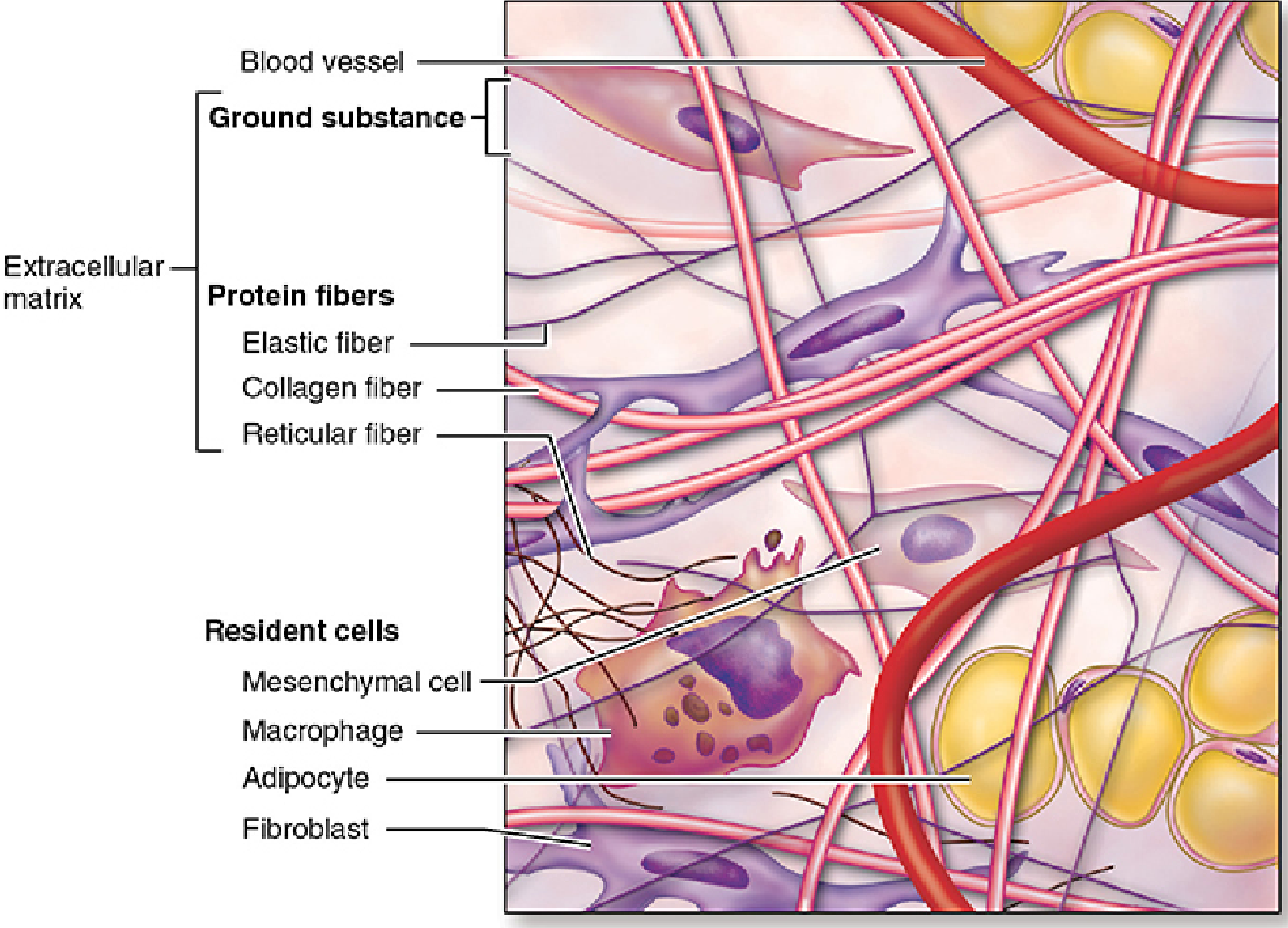
Part 1 - Types of Connective Tissue
A. Embryonic Connective Tissues
| Type | Structure | Function | Location |
|---|---|---|---|
| Mesenchyme | Sparse, undifferentiated spindle cells in sparse collagen matrix | Contains stem/progenitor cells for all adult connective tissue | Mesodermal layer of early embryo |
| Mucoid (Mucous) Connective Tissue | Random fibroblasts and collagen fibers in viscous hyaluronan-rich matrix | Supports and cushions large blood vessels | Wharton's jelly of fetal umbilical cord |
B. Connective Tissue Proper
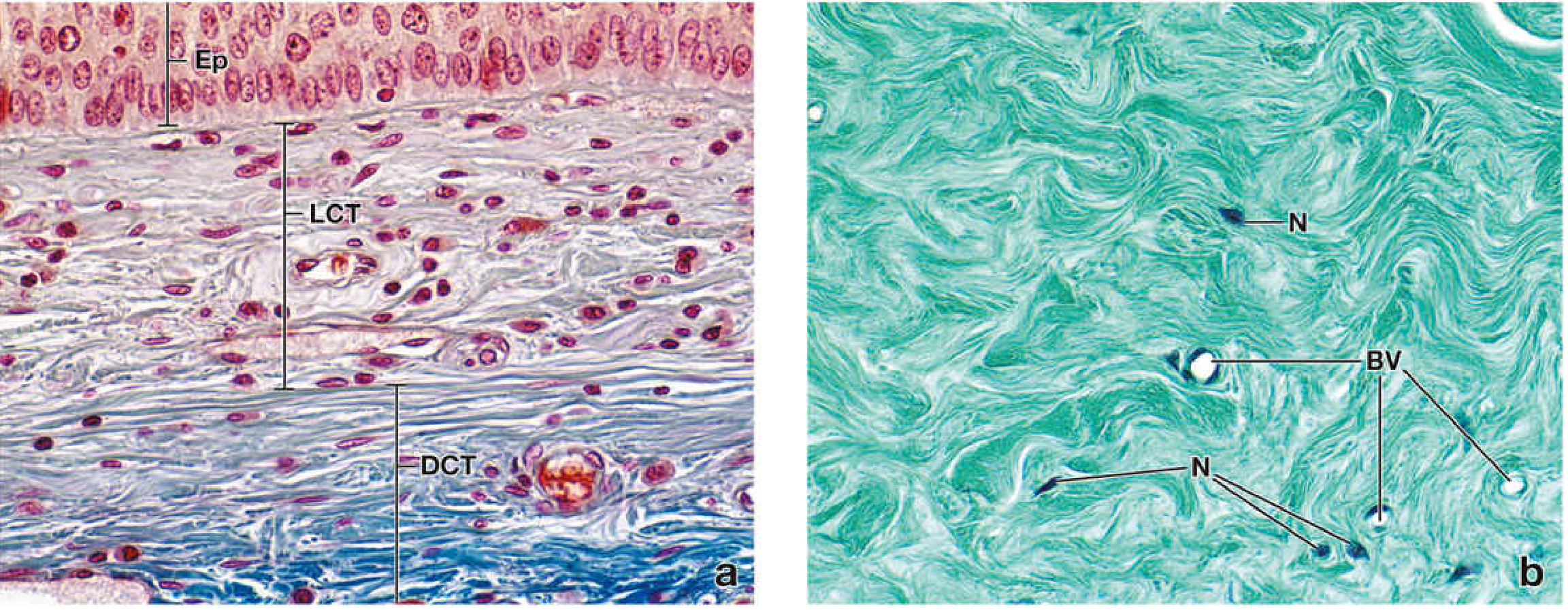
1. Loose (Areolar) Connective Tissue
- Structure: Abundant ground substance; moderate collagen (randomly arranged) and elastic fibers; many diverse cell types
- Characteristics: Cells, fibers, and ground substance in roughly equal proportions; delicate, flexible; appears pale/loosely organized on H&E
- Function: Supports microvasculature, nerves, and immune defense cells; medium for diffusion of nutrients/waste; acts as the battleground for immune responses
- Locations: Lamina propria beneath GI and respiratory epithelium; adventitia of blood vessels; dermis (superficial layer); mesenteries; fill spaces between other tissue types
2. Dense Irregular Connective Tissue
- Structure: Predominantly thick type I collagen bundles arranged randomly in multiple directions; few cells (mainly fibroblasts); little ground substance
- Characteristics: Collagen bundles interweave in all planes, resisting forces from multiple directions; strongly eosinophilic on H&E
- Function: Protects and supports organs; resists tearing and stretching from all directions
- Locations: Dermis of skin, organ capsules (kidney, testis, spleen, lymph nodes), submucosa of GI tract, periosteum, perichondrium
3. Dense Regular Connective Tissue
- Structure: Tightly packed parallel bundles of type I collagen with fibroblasts (tenocytes) aligned between bundles; minimal ground substance; almost no other cell types
- Characteristics: Glistening white, inextensible; extremely strong in one direction
- Function: Transmits forces; provides strong connections within the musculoskeletal system; resists tensile forces along the axis of fibers
- Locations: Tendons (muscle to bone), ligaments (bone to bone), aponeuroses, corneal stroma (collagen layers orthogonally arranged for transparency)
C. Specialized Connective Tissues
| Type | Key Features | Location |
|---|---|---|
| Reticular Connective Tissue | Delicate type III collagen (reticulin) network; argyrophilic (silver-staining); produced by reticular cells | Bone marrow, liver, spleen, lymph nodes, all lymphoid organs, endocrine glands |
| Adipose Tissue | Dominated by adipocytes; ECM is sparse; subdivided into unilocular (white) and multilocular (brown) | Subcutaneous tissue, omentum, bone marrow, periorbital |
| Cartilage | Rigid ECM rich in type II collagen and proteoglycans; avascular; chondrocytes in lacunae | Articular surfaces, trachea, ear, nose, intervertebral discs |
| Bone | Mineralized ECM (hydroxyapatite); type I collagen; osteocytes in lacunae | Skeleton |
| Blood | Liquid ECM (plasma); circulating cells | Cardiovascular system |
Part 2 - Functions of Connective Tissue
| Function | Mechanism |
|---|---|
| Structural support | ECM fibers provide tensile strength and framework for all tissues and organs |
| Binding and connection | Connects epithelia to underlying structures; binds muscles to bone (tendons), bone to bone (ligaments) |
| Metabolic exchange | Interstitial fluid within ground substance serves as the direct medium for diffusion of O₂, nutrients, CO₂, and metabolic wastes between cells and blood |
| Immune defense | Contains macrophages, lymphocytes, plasma cells, neutrophils, eosinophils, and mast cells that survey and respond to pathogens and foreign material |
| Energy storage | Adipocytes store neutral lipids; mobilized as fatty acids during energy demands |
| Transport | Ground substance is the medium through which substances travel from blood to parenchymal cells |
| Tissue repair and regeneration | Fibroblasts proliferate and deposit new ECM after injury; wound healing |
| Scaffolding for organogenesis | Provides the framework on which developing organs are shaped |
| Thermoregulation | Brown adipose tissue generates heat via uncoupling of oxidative phosphorylation |
Part 3 - Cells of Connective Tissue
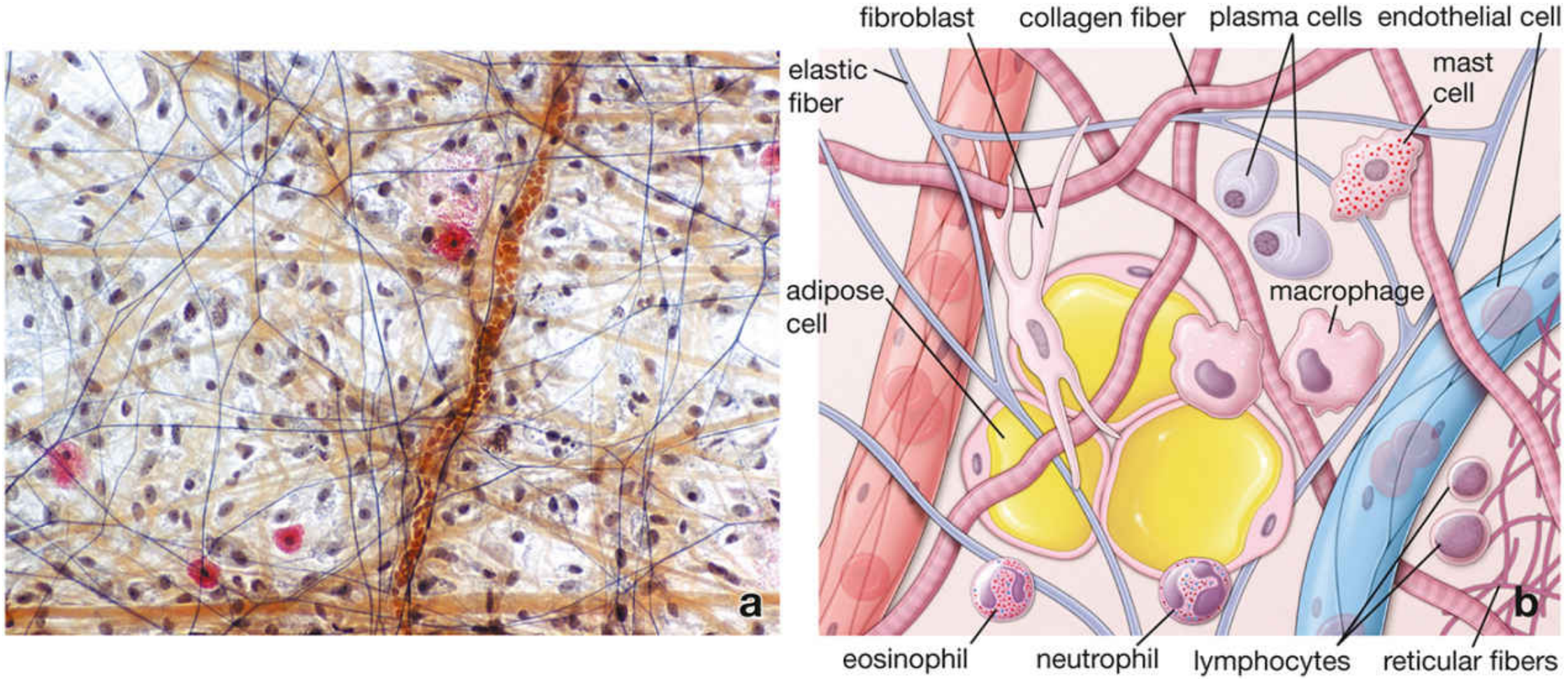
Resident (Permanent) Cells
1. Fibroblasts / Fibrocytes
- Most common cell in connective tissue proper
- Origin: Local mesenchymal cells; permanent residents
- Morphology: Elongated, spindle-shaped with pale, euchromatic nucleus and inconspicuous nucleoli (fibrocyte = inactive form); abundant RER and large Golgi when active (fibroblast)
- Function: Synthesize and secrete ALL ECM components:
- Collagen (most abundant body protein)
- Elastin
- GAGs, proteoglycans
- Multiadhesive glycoproteins (fibronectin, laminin)
- Ground substance
- Key role: Tissue maintenance and wound repair (fibroblasts at wound sites activate, proliferate, and form granulation tissue)
- Note: In specific locations, fibroblasts have specialized names: tenocytes (tendons), chondroblasts (cartilage), osteoblasts (bone), odontoblasts (teeth)
2. Adipocytes
- Unilocular (white) adipocytes: Single large lipid droplet; nucleus displaced to periphery; "signet ring" appearance on H&E
- Multilocular (brown) adipocytes: Multiple small lipid droplets; centrally placed nucleus; rich in mitochondria
- Function: Energy storage, thermal insulation, cushioning, endocrine functions (leptin, adiponectin secretion)
3. Mast Cells
- Origin: Bone marrow progenitors; mature in connective tissue
- Morphology: Large ovoid cells; metachromatic granules (stain purple with toluidine blue); bilobed or round nucleus
- Location: Especially numerous near small blood vessels in skin, mesenteries, and mucous membranes (strategic sentinel positions)
- Granule contents and secretions:
- Histamine: Vasodilation, increased vascular permeability
- Heparin: Anticoagulation
- Tryptase and chymase: Serine proteases; tissue remodeling
- Eosinophil and neutrophil chemotactic factors: Recruit leukocytes
- Cytokines: Direct leukocyte activities
- Phospholipid precursors: Converted to prostaglandins and leukotrienes
- Function: Immediate hypersensitivity (IgE-mediated); primary mediators of allergic reactions and anaphylaxis; inflammation and defense against parasites
4. Macrophages (Histiocytes)
- Origin: Bone marrow monocytes → enter blood → migrate into connective tissue → differentiate into macrophages
- Morphology: Irregular, pleomorphic cells; kidney-shaped nucleus; extensive pseudopods; lysosomes visible on EM
- Function:
- Phagocytosis of bacteria, debris, worn-out cells, ECM components
- Antigen processing and presentation to T lymphocytes (MHC class II)
- Secretion of cytokines (IL-1, TNF-α, IL-6), growth factors (TGF-β, FGF), and enzymes (collagenase)
- Tissue homeostasis and repair coordination
- Specialized macrophage forms: Kupffer cells (liver), microglia (brain), osteoclasts (bone), alveolar macrophages (lung), Langerhans cells (skin)
Transient (Immigrant) Cells - Arrive from Blood as Needed
5. Plasma Cells
- Origin: Differentiated B lymphocytes
- Morphology: Large, ovoid; basophilic cytoplasm rich in RER; large Golgi (pale perinuclear zone); "clock-face" or "cartwheel" heterochromatin pattern in nucleus
- Function: Produce and secrete immunoglobulins (antibodies)
- Locations: Abundant in connective tissue of GI tract (lamina propria), respiratory tract, salivary glands - sites of antigen exposure
6. Lymphocytes
- Origin: Bone marrow; circulate in blood and lymph
- Morphology: Small, round cells with large, dark nucleus and scant cytoplasm
- Types: T cells (cell-mediated immunity), B cells (antibody production), NK cells
- Function: Adaptive immune surveillance; coordinate immune responses
7. Neutrophilic Leukocytes
- Origin: Bone marrow; short-lived (days)
- Morphology: Multilobed nucleus (3-5 lobes); granules contain lysozyme, defensins, myeloperoxidase
- Function: First responders; phagocytosis of bacteria; primary cells of acute inflammation
8. Eosinophilic Leukocytes
- Morphology: Bilobed nucleus; large eosinophilic (pink) granules containing major basic protein, eosinophil peroxidase
- Function: Modulate allergic/IgE reactions; defense against parasites; phagocytosis of antigen-antibody complexes
9. Basophilic Leukocytes / Basophils
- Morphology: Segmented nucleus obscured by dark basophilic granules
- Function: Similar to mast cells - release histamine and heparin; participate in IgE-mediated reactions
| Cell | Origin | Key Function |
|---|---|---|
| Fibroblast/Fibrocyte | Mesenchyme | ECM synthesis and maintenance |
| Adipocyte | Mesenchyme | Fat storage, endocrine |
| Mast cell | Bone marrow | Allergy, inflammation, defense |
| Macrophage | Bone marrow monocyte | Phagocytosis, antigen presentation, cytokines |
| Plasma cell | B lymphocyte | Antibody secretion |
| Lymphocyte | Bone marrow | Immune surveillance |
| Neutrophil | Bone marrow | Bacterial phagocytosis |
| Eosinophil | Bone marrow | Anti-parasite, allergy modulation |
Part 4 - Fibers of Connective Tissue
1. Collagen Fibers
Structure and Synthesis
- Basic unit: procollagen, a triple helix of three α-chains, each ~300 nm long, 1.5 nm wide; every third amino acid is glycine (Gly-X-Y repeat)
- Synthesis steps: polyribosome assembly in RER → hydroxylation of proline and lysine (requires Vitamin C) → glycosylation → triple helix formation → secretion → extracellular cleavage of propeptides → self-assembly into fibrils → cross-linking by lysyl oxidase
- Vitamin C deficiency impairs proline/lysine hydroxylation → unstable triple helix → scurvy (friable collagen)
Collagen Types and Their Locations
| Type | Fiber Structure | Major Locations |
|---|---|---|
| Type I | Thick fibers/bundles; most abundant (90% of all collagen) | Bone, dermis, tendons, ligaments, organ capsules, cornea, fascia |
| Type II | Thin fibrils | Hyaline and elastic cartilage, vitreous humor of eye, nucleus pulposus |
| Type III | Thin reticular fibers; forms reticulin | Lymphoid organs, bone marrow, liver, lung, blood vessels (early wound healing) |
| Type IV | Non-fibrillar sheet; meshwork | Basal laminae (basement membranes) of all epithelia |
| Type V | Mixed with type I | Cornea, bone, placenta |
| Type VII | Anchoring fibrils | Skin - anchors epidermis to dermis |
| Type IX, X, XI | Fibril-associated collagens | Cartilage matrix regulation |
Histological Staining of Collagen
- H&E: eosinophilic (pink)
- Masson's trichrome: blue or green
- PAS: weakly positive (due to glycosylation)
- Gomori silver: highlights type III (reticular fibers turn black)
2. Reticular Fibers
- Composed of type III collagen with high carbohydrate content (~10% vs. 1% in type I)
- Very thin (0.5-2 μm diameter); form delicate branching networks (reticula)
- Staining: Argyrophilic (black with silver impregnation); PAS-positive; poorly visible with H&E
- Produced by: Fibroblasts (called reticular cells in hematopoietic/lymphoid organs)
- Function: Supportive scaffold for rapidly dividing and phagocytic cells; stroma support for secretory parenchyma
3. Elastic Fibers
- Provide elasticity and resilience - allow tissue to stretch and recoil
- Thinner than type I collagen fibers; form sparse interspersed networks or (in large arteries) fenestrated elastic lamellae
- Composition: Core of elastin (60 kDa; cross-linked via desmosine rings formed from lysine residues) surrounded by a sheath of fibrillin microfibrils (10 nm diameter)
- Cross-linking: Lysyl oxidase converts lysine amino groups to aldehydes; four oxidized lysines condense as desmosine rings - provides the rubbery, reversible extensibility
- Synthesis: Fibroblasts (and smooth muscle cells in vessel walls) secrete fibrillin microfibrils first (scaffold), then deposit elastin around them
- H&E: Not strongly acidophilic; hard to see
- Orcein or aldehyde fuchsin: Dark brown-black staining
- Weigert's elastic stain: Blue-black
| Feature | Collagen (Type I) | Reticular (Type III) | Elastic |
|---|---|---|---|
| Protein | Collagen I | Collagen III | Elastin + fibrillin |
| Diameter | 1-20 μm (bundles) | 0.5-2 μm | 0.2-1 μm |
| Arrangement | Parallel or random bundles | Delicate networks | Branching networks or lamellae |
| H&E stain | Pink | Poorly visible | Pale/barely visible |
| Special stain | Masson trichrome (blue) | Silver stain (black) | Orcein (dark) |
| Strength | High tensile strength | Delicate support | Elastic recoil |
Part 5 - Extracellular Matrix (ECM)
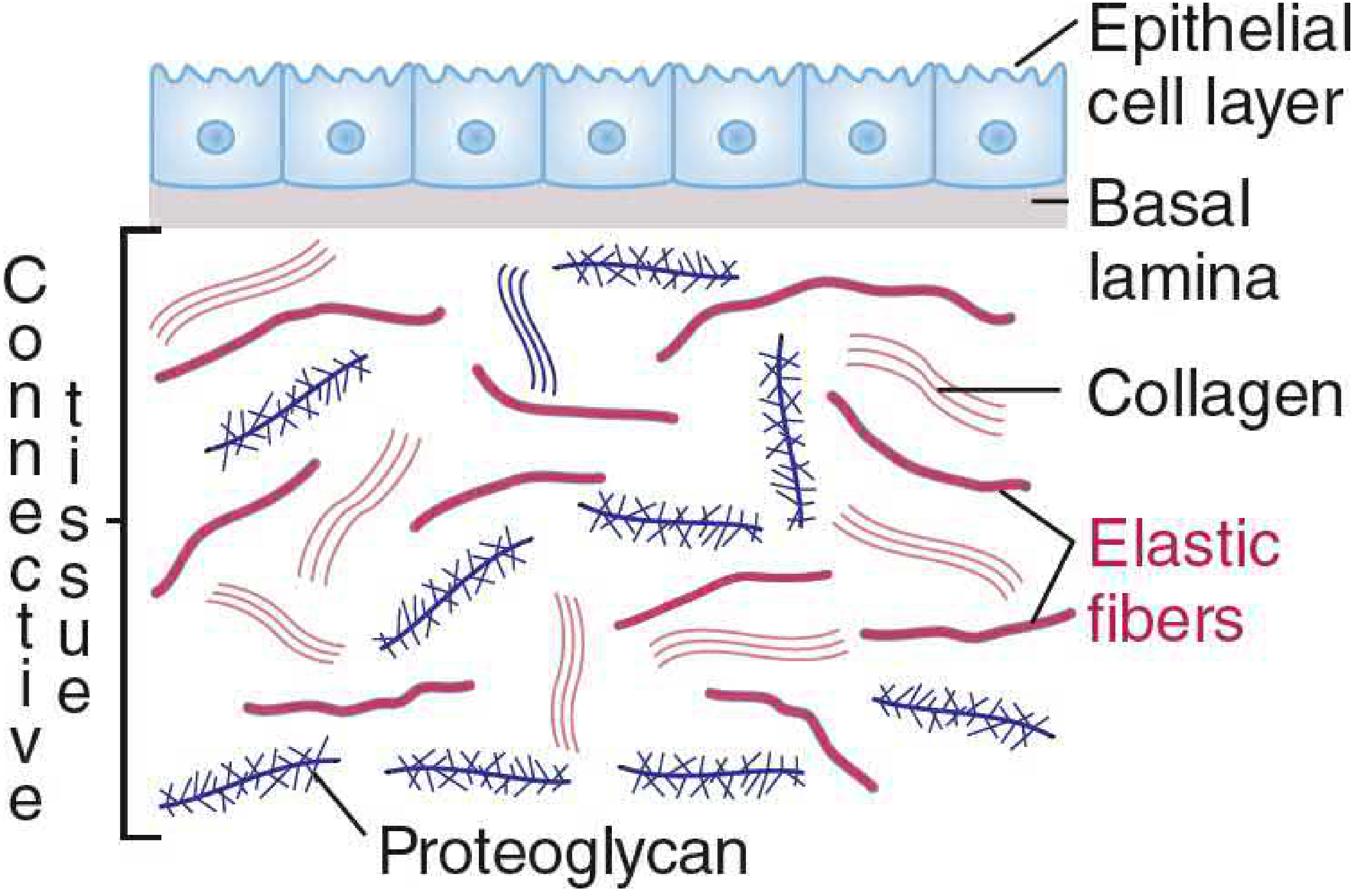
Ground Substance
A. Glycosaminoglycans (GAGs)
| GAG | Key Features | Location |
|---|---|---|
| Hyaluronan (Hyaluronic acid) | Largest GAG; not sulfated; not covalently bound to protein; extremely hydrophilic | Ubiquitous: synovial fluid, vitreous humor, umbilical cord, embryonic tissues |
| Chondroitin sulfate | Sulfated; most abundant GAG in body | Cartilage, bone, tendon, skin |
| Dermatan sulfate | Sulfated; associated with type I collagen | Skin, blood vessels, heart valves, tendons |
| Heparan sulfate | Sulfated; component of basal laminae | Basement membranes, cell surfaces |
| Heparin | Highly sulfated; stored in mast cell granules | Mast cells, anticoagulation |
| Keratan sulfate | Sulfated; lacks uronic acid | Cornea, cartilage, intervertebral disc |
B. Proteoglycans
- Aggrecan: Large cartilage proteoglycan; binds hyaluronan + chondroitin sulfate; responsible for cartilage's compressive resistance
- Perlecan: Major heparan sulfate proteoglycan of basement membranes
- Decorin, Biglycan: Small leucine-rich proteoglycans; regulate collagen fibrillogenesis
- Syndecan: Cell surface proteoglycan; functions as co-receptor
C. Multiadhesive Glycoproteins
| Glycoprotein | Binds to | Function |
|---|---|---|
| Fibronectin | Collagen, heparan sulfate, integrins, fibrin | Cell adhesion, migration, wound healing; essential for fibroblast and immune cell movement through matrix |
| Laminin | Type IV collagen, nidogen, perlecan, integrins | Major glycoprotein of basal laminae; promotes cell attachment, differentiation, and polarization |
| Nidogen/Entactin | Laminin + type IV collagen | Bridging molecule linking the two lamina components |
| Tenascin | Fibronectin, proteoglycans | Anti-adhesive; modulates cell-matrix interactions; expressed in wound healing and tumor stroma |
| SPARC (Osteopontin) | Collagen, integrins | Regulates ECM assembly and cell-matrix signaling |
D. Cell-Matrix Interaction: Integrins
- Integrins are transmembrane receptors that bind ECM glycoproteins (fibronectin, laminin, collagen) on the outside and link to the actin cytoskeleton (via talin, vinculin) on the inside
- They transduce mechanical and biochemical signals bidirectionally between ECM and cell interior
- Critical for cell migration, differentiation, proliferation, and survival
E. Matrix Metalloproteinases (MMPs) and TIMPs
- MMPs: Enzymes that degrade ECM components (collagenases, gelatinases, stromelysins); required for cell migration, tissue remodeling, and repair
- TIMPs (Tissue inhibitors of MMPs): Regulate MMP activity
- Clinical: Dysregulation allows cancer cells to invade and metastasize; also implicated in arthritis, aneurysms, and fibrosis
Part 6 - Summary of Connective Tissue Types with Locations
| CT Type | Key ECM Feature | Cells Present | Body Location |
|---|---|---|---|
| Loose (areolar) | Abundant ground substance, mixed loose fibers | Fibroblasts, macrophages, mast cells, plasma cells, leukocytes | Lamina propria (GI, respiratory tracts), dermis, mesenteries, nerve/muscle sheaths |
| Dense irregular | Thick collagen bundles, random arrangement | Mainly fibroblasts | Skin dermis, organ capsules (kidney, testis), submucosa, periosteum |
| Dense regular | Parallel collagen bundles | Fibroblasts (tenocytes) | Tendons, ligaments, aponeuroses, corneal stroma |
| Reticular | Type III collagen network | Reticular cells (fibroblasts) | Bone marrow, liver, spleen, lymph nodes, endocrine glands |
| Adipose (white) | Minimal ECM; unilocular adipocytes dominate | Adipocytes | Subcutaneous fat, omentum, retroperitoneum, breast |
| Adipose (brown) | Minimal ECM; multilocular adipocytes | Adipocytes | Interscapular region (infants), perirenal, neck |
| Hyaline cartilage | Type II collagen + aggrecan | Chondrocytes | Articular surfaces, trachea, bronchi, costal cartilage, larynx |
| Elastic cartilage | Type II collagen + elastic fibers | Chondrocytes | Ear pinna, epiglottis, auditory tube |
| Fibrocartilage | Mostly type I collagen | Chondrocytes | Intervertebral discs, pubic symphysis, menisci |
| Bone (compact) | Mineralized type I collagen in osteons | Osteocytes, osteoblasts, osteoclasts | Diaphysis of long bones, flat bones |
| Bone (spongy) | Mineralized type I collagen; trabeculae | Same as compact | Epiphyses, vertebral bodies, diploe of flat bones |
| Blood | Plasma (liquid ECM) | RBCs, WBCs, platelets | Cardiovascular system |
| Mucoid | Hyaluronan-rich viscous matrix | Fibroblasts | Umbilical cord (Wharton's jelly) |
| Mesenchyme | Sparse collagen; viscous proteoglycan matrix | Mesenchymal stem cells | Embryo |
- Junqueira's Basic Histology - Text and Atlas, 17th Edition - Chapters 5 (Connective Tissue Proper)
- Ross Histology - A Text and Atlas with Correlated Cell and Molecular Biology - Chapter 6 (Connective Tissue)
- Basic Medical Biochemistry: A Clinical Approach, 6th Edition - Chapter 47 (Extracellular Matrix and Connective Tissue)
Create a summary PDF comparing all CT types with histology images
mkdir -p /home/daytona/workspace/connective-tissue-pdf && ls /home/daytona/workspace/
~/connective-tissue-pdf/build_pdf.py
#!/usr/bin/env python3
"""
Build a comprehensive Connective Tissue comparison PDF with histology images.
"""
import urllib.request
import os
import io
from reportlab.lib.pagesizes import A4
from reportlab.lib import colors
from reportlab.lib.styles import getSampleStyleSheet, ParagraphStyle
from reportlab.lib.units import cm, mm
from reportlab.platypus import (
SimpleDocTemplate, Paragraph, Spacer, Table, TableStyle,
Image, PageBreak, HRFlowable, KeepTogether
)
from reportlab.platypus.flowables import HRFlowable
from reportlab.lib.enums import TA_CENTER, TA_LEFT, TA_JUSTIFY
from reportlab.pdfbase import pdfmetrics
from reportlab.pdfbase.ttfonts import TTFont
# ── Output path ────────────────────────────────────────────────────────────────
OUTPUT = "/home/daytona/workspace/connective-tissue-pdf/Connective_Tissue_Summary.pdf"
IMG_DIR = "/home/daytona/workspace/connective-tissue-pdf/images"
os.makedirs(IMG_DIR, exist_ok=True)
# ── Image URLs (from Junqueira's + Ross Histology textbooks) ──────────────────
IMAGES = {
"ct_components": "https://cdn.orris.care/cdss_images/00c882cccb96ef3d8ba8a0692508941fe3abcd7bf4541485469b212df382324e.png",
"loose_dense_histo": "https://cdn.orris.care/cdss_images/296617e98af409f96cb62ba5366913cd0185e7cb888ea3b03b407ecfdaed61a8.png",
"loose_ct_diagram": "https://cdn.orris.care/cdss_images/3519723abb817746727ef1a5567889d7244a66e15ca00966b8fec139151f2327.png",
"loose_ct_section": "https://cdn.orris.care/cdss_images/3d89ba8e1f0f6e698d8d155713d82030d1ea02e24a7ab389c7db7eb7697741a7.png",
"dense_irregular": "https://cdn.orris.care/cdss_images/b6bd8d7a398ea733232bae89b74a6390f768b9bb191c2ffea42b239bd50d8ae6.png",
"tendon_longitudinal": "https://cdn.orris.care/cdss_images/9c70ef39f4ed67ed512c16f75c01ddd2ea4da49785ba35369e277cf327a35152.png",
"reticular_fibers_1": "https://cdn.orris.care/cdss_images/b769964d13f7fb39002a12dd72cd1cf744a21171b8496c40cc67a0f6da148502.png",
"reticular_fibers_2": "https://cdn.orris.care/cdss_images/f091cb15dfdc09138c9a732bdd4090c0a724e8305b01ab423852a8866308edfb.png",
"elastic_fibers": "https://cdn.orris.care/cdss_images/f823c0233b3b6128b63f20f5172bbbb7fad7a97fa13cd1fec665da497d61dbc6.png",
"collagen_triple_helix": "https://cdn.orris.care/cdss_images/edb4b159545616a2a56fca8ee0d33e0f29f2161bfbc28b63f45210336d469f2f.png",
"ecm_overview": "https://cdn.orris.care/cdss_images/587dc018680ba9587b33609fa74007b74bfa1dae94c0a90c1f1d1eed79815f01.png",
"mesenchyme": "https://cdn.orris.care/cdss_images/c787a22d227a732e462532e6bbea76b7e6b627e90ccf952cace14d0736ccd221.png",
"lv_hypertrophy_normal": "https://cdn.orris.care/cdss_images/d52449c76c8b9dabef9b79bf6f5f4952f0c87432de5573454f25676dcabe9de0.png", # normal vs hypertrophied myocardium (histology comparison)
}
def download_image(name, url):
path = os.path.join(IMG_DIR, f"{name}.png")
if not os.path.exists(path):
try:
req = urllib.request.Request(url, headers={"User-Agent": "Mozilla/5.0"})
with urllib.request.urlopen(req, timeout=15) as r:
data = r.read()
with open(path, "wb") as f:
f.write(data)
print(f" ✓ {name}")
except Exception as e:
print(f" ✗ {name}: {e}")
return None
return path
print("Downloading images...")
img_paths = {k: download_image(k, v) for k, v in IMAGES.items()}
print("Done.\n")
# ── Styles ────────────────────────────────────────────────────────────────────
styles = getSampleStyleSheet()
def make_style(name, parent="Normal", **kw):
s = ParagraphStyle(name, parent=styles[parent], **kw)
return s
title_style = make_style("DocTitle", "Heading1", fontSize=22, textColor=colors.HexColor("#1a3d6b"),
spaceAfter=6, alignment=TA_CENTER, fontName="Helvetica-Bold")
subtitle_style= make_style("DocSubtitle", "Normal", fontSize=11, textColor=colors.HexColor("#4a6fa5"),
spaceAfter=16, alignment=TA_CENTER)
h1_style = make_style("H1", "Heading1", fontSize=16, textColor=colors.HexColor("#1a3d6b"),
spaceBefore=14, spaceAfter=6, fontName="Helvetica-Bold",
borderPad=4, backColor=colors.HexColor("#e8f0f9"),
borderColor=colors.HexColor("#1a3d6b"), borderWidth=0.5)
h2_style = make_style("H2", "Heading2", fontSize=13, textColor=colors.HexColor("#2c5f8a"),
spaceBefore=10, spaceAfter=4, fontName="Helvetica-Bold")
h3_style = make_style("H3", "Heading3", fontSize=11, textColor=colors.HexColor("#2c5f8a"),
spaceBefore=8, spaceAfter=3, fontName="Helvetica-BoldOblique")
body_style = make_style("Body", "Normal", fontSize=9.5, leading=14,
spaceAfter=4, alignment=TA_JUSTIFY)
caption_style = make_style("Caption", "Normal", fontSize=8, textColor=colors.HexColor("#555555"),
alignment=TA_CENTER, spaceAfter=8, fontName="Helvetica-Oblique")
bullet_style = make_style("Bullet", "Normal", fontSize=9.5, leading=13,
leftIndent=12, spaceAfter=2)
source_style = make_style("Source", "Normal", fontSize=7.5, textColor=colors.HexColor("#777777"),
alignment=TA_CENTER, spaceAfter=2)
toc_style = make_style("TOC", "Normal", fontSize=10, leading=16, leftIndent=8)
PAGE_W, PAGE_H = A4
def img_flowable(key, width_cm, caption=""):
p = img_paths.get(key)
if not p or not os.path.exists(p):
return Spacer(1, 0.3*cm)
w = width_cm * cm
items = [Image(p, width=w, height=None)] # auto height
if caption:
items.append(Paragraph(caption, caption_style))
return items
# ── Helper: colored section header ───────────────────────────────────────────
def section_header(text, color="#1a3d6b", bg="#e8f0f9"):
data = [[Paragraph(text, make_style("SH", "Normal", fontSize=13,
fontName="Helvetica-Bold",
textColor=colors.HexColor(color)))]]
t = Table(data, colWidths=[17*cm])
t.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), colors.HexColor(bg)),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING", (0,0), (-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 10),
("RIGHTPADDING", (0,0), (-1,-1), 6),
("LINEBELOW", (0,0), (-1,-1), 1.5, colors.HexColor(color)),
]))
return t
# ── Build document ─────────────────────────────────────────────────────────────
def build():
doc = SimpleDocTemplate(
OUTPUT,
pagesize=A4,
leftMargin=2*cm, rightMargin=2*cm,
topMargin=2.2*cm, bottomMargin=2.2*cm,
title="Connective Tissue – Complete Summary",
author="Medical Library (Junqueira's, Ross Histology)",
)
story = []
W = 17*cm # usable width
# ── COVER ──────────────────────────────────────────────────────────────────
story += [
Spacer(1, 1*cm),
Paragraph("CONNECTIVE TISSUE", title_style),
Paragraph("Types · Functions · Cells · Fibers · ECM · Locations", subtitle_style),
HRFlowable(width=W, thickness=2, color=colors.HexColor("#1a3d6b")),
Spacer(1, 0.4*cm),
]
# Cover diagram
for fl in img_flowable("ct_components", 14,
"Fig. 1 – Connective tissue components: collagen fibers (pink), elastic fibers (thin branching),\n"
"reticular fibers (fine), and ground substance with resident cells.\n"
"(Junqueira's Basic Histology, 17e)"):
story.append(fl)
story += [
Spacer(1, 0.5*cm),
Paragraph(
"Connective tissue (CT) forms the structural framework of the body. Unlike epithelium, muscle, "
"and nerve – which are cell-dominant – CT is characterized by a large extracellular matrix (ECM) "
"that exceeds the cellular volume in virtually all subtypes. All CT originates from embryonic "
"<b>mesenchyme</b> (mesoderm-derived). The diversity of CT types reflects varying compositions "
"of cells, fibers, and ground substance.",
body_style),
Spacer(1, 0.3*cm),
Paragraph("<b>Sources:</b> Junqueira's Basic Histology 17e · Ross Histology 9e · Basic Medical Biochemistry 6e",
source_style),
PageBreak(),
]
# ── SECTION 1 – CLASSIFICATION TABLE ──────────────────────────────────────
story.append(section_header("1. Classification of Connective Tissue"))
story.append(Spacer(1, 0.3*cm))
# Main classification table
header = ["CT Type", "ECM / Structure", "Cells", "Key Functions", "Body Locations"]
rows = [
# Embryonic
["Mesenchyme\n(Embryonic)", "Sparse collagen; viscous hyaluronan-rich matrix; few fibers",
"Undifferentiated mesenchymal stem cells", "Progenitor of all adult CT cells",
"Embryonic mesoderm"],
["Mucoid / Mucous\n(Embryonic)", "Abundant hyaluronan; viscous gel; sparse collagen",
"Fibroblasts", "Cushions and supports umbilical vessels",
"Wharton's jelly\n(umbilical cord)"],
# Connective tissue proper
["Loose (Areolar)\nCT Proper", "Equal parts cells, random collagen & elastic fibers, abundant ground substance",
"Fibroblasts, macrophages, mast cells, plasma cells, leukocytes, adipocytes",
"Metabolic exchange; immune defense; flexible support; microvasculature support",
"Lamina propria (GI, respiratory); dermis; mesenteries; perivascular sheaths"],
["Dense Irregular\nCT Proper", "Thick type I collagen bundles in random multi-directional arrangement; little ground substance",
"Mainly fibroblasts", "Resist tearing from all directions; protect & support organs",
"Skin dermis; organ capsules (kidney, testis, spleen); submucosa; periosteum"],
["Dense Regular\nCT Proper", "Parallel packed type I collagen bundles; minimal ground substance",
"Fibroblasts (tenocytes/tendinocytes)", "Transmit tensile forces; strong unidirectional connections",
"Tendons, ligaments, aponeuroses, corneal stroma"],
# Specialized
["Reticular CT\n(Specialized)", "Delicate type III collagen (reticulin) network; argyrophilic",
"Reticular cells (specialized fibroblasts)", "Stroma for lymphoid/hematopoietic cells",
"Bone marrow, liver, spleen, lymph nodes, endocrine glands"],
["Adipose – White\n(Specialized)", "Sparse ECM; unilocular adipocytes dominate",
"White adipocytes (single large lipid droplet)", "Energy storage; insulation; cushioning; leptin secretion",
"Subcutaneous fat, omentum, retroperitoneum, breast, bone marrow"],
["Adipose – Brown\n(Specialized)", "Sparse ECM; multilocular adipocytes; mitochondria-rich",
"Brown adipocytes (multiple lipid droplets)", "Non-shivering thermogenesis (UCP-1)",
"Interscapular (infants), perirenal, neck"],
["Hyaline Cartilage\n(Specialized)", "Type II collagen + aggrecan proteoglycans; avascular; glassy matrix",
"Chondrocytes in lacunae", "Smooth articulation; resist compression; framework",
"Articular surfaces, trachea, costal cartilage, larynx, bronchi"],
["Elastic Cartilage\n(Specialized)", "Type II collagen + abundant elastic fibers",
"Chondrocytes", "Flexible support; maintains shape after deformation",
"Ear pinna, epiglottis, auditory tube"],
["Fibrocartilage\n(Specialized)", "Predominantly type I collagen; densely packed fibers",
"Chondrocytes in rows", "Resist compression and shear forces; shock absorption",
"Intervertebral discs, pubic symphysis, menisci, TMJ disc"],
["Compact Bone\n(Specialized)", "Mineralized type I collagen; hydroxyapatite; Haversian system (osteons)",
"Osteocytes, osteoblasts, osteoclasts", "Rigid support; leverage for movement; Ca²⁺ reservoir",
"Diaphysis of long bones, outer layer of flat bones"],
["Spongy Bone\n(Specialized)", "Mineralized type I collagen; trabecular lattice with marrow spaces",
"Same as compact bone", "Lightweight strength; houses red marrow (hematopoiesis)",
"Epiphyses, vertebral bodies, diploe of flat bones"],
["Blood\n(Specialized)", "Liquid plasma (90% water, proteins, ions) = liquid ECM",
"Erythrocytes, leukocytes, platelets", "O₂/CO₂ transport; immunity; hemostasis",
"Cardiovascular system (circulation)"],
]
# Style the table
col_widths = [3.0*cm, 3.8*cm, 3.0*cm, 3.5*cm, 3.7*cm]
all_data = [
[Paragraph(f"<b>{h}</b>", make_style("TH","Normal",fontSize=8.5,
fontName="Helvetica-Bold", textColor=colors.white,
alignment=TA_CENTER)) for h in header]
]
for i, row in enumerate(rows):
bg = colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white
all_data.append([Paragraph(c, make_style(f"TC{i}","Normal",fontSize=8,
leading=11)) for c in row])
tbl = Table(all_data, colWidths=col_widths, repeatRows=1)
tbl_style = TableStyle([
# Header row
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#1a3d6b")),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
("FONTNAME", (0, 0), (-1, 0), "Helvetica-Bold"),
("FONTSIZE", (0, 0), (-1, 0), 8.5),
("ALIGN", (0, 0), (-1, 0), "CENTER"),
# Alternating row colours
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(rows))],
# Grid
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 4),
("BOTTOMPADDING",(0, 0), (-1, -1), 4),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
# Section dividers (embryonic / CT proper / specialized)
("LINEABOVE", (0, 1), (-1, 1), 1.2, colors.HexColor("#2c5f8a")), # after header
("LINEABOVE", (0, 3), (-1, 3), 1.2, colors.HexColor("#2c5f8a")), # CT Proper starts
("LINEABOVE", (0, 6), (-1, 6), 1.2, colors.HexColor("#2c5f8a")), # Specialized starts
# First column bold
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
("FONTSIZE", (0, 1), (0, -1), 8),
])
tbl.setStyle(tbl_style)
story.append(tbl)
story.append(Spacer(1, 0.5*cm))
# Section labels
legend_data = [
[Paragraph("<b>Shading guide:</b>", body_style),
Paragraph("Rows 1–2: Embryonic CT | Rows 3–5: Connective Tissue Proper | Rows 6–14: Specialized CT",
make_style("leg","Normal",fontSize=8, textColor=colors.HexColor("#444444")))],
]
legend = Table(legend_data, colWidths=[3.5*cm, 13.5*cm])
legend.setStyle(TableStyle([("TOPPADDING",(0,0),(-1,-1),2),("BOTTOMPADDING",(0,0),(-1,-1),2)]))
story.append(legend)
story.append(PageBreak())
# ── SECTION 2 – HISTOLOGY IMAGES ──────────────────────────────────────────
story.append(section_header("2. Histology of Connective Tissue Types"))
story.append(Spacer(1, 0.4*cm))
# ── 2a Loose CT ──────────────────────────────────────────────────────────
story.append(Paragraph("2a. Loose (Areolar) Connective Tissue", h2_style))
story.append(Paragraph(
"Loose CT is the most widely distributed CT in the body. It contains cells, collagen fibers, "
"elastic fibers, and reticular fibers in roughly equal proportions, with abundant ground "
"substance (hydrated proteoglycans). It is flexible and metabolically active, housing immune "
"cells that survey for pathogens. Fibroblasts are the dominant cell type.",
body_style))
# Two images side by side: photomicrograph + diagram
loose_img_path = img_paths.get("loose_ct_section")
loose_diag_path = img_paths.get("loose_ct_diagram")
img_row_data = [[]]
img_row_captions = [[]]
if loose_img_path and os.path.exists(loose_img_path):
img_row_data[0].append(Image(loose_img_path, width=8*cm, height=6*cm))
img_row_captions[0].append(Paragraph(
"H&E: Loose CT (L) with fine fibers and\nmany varied nuclei; Dense CT (D) below\n(Junqueira's 17e)",
caption_style))
if loose_diag_path and os.path.exists(loose_diag_path):
img_row_data[0].append(Image(loose_diag_path, width=8*cm, height=6*cm))
img_row_captions[0].append(Paragraph(
"Diagram of loose CT components:\nfibroblast, macrophage, mast cell,\nplasma cell, all fiber types\n(Ross Histology 9e)",
caption_style))
if img_row_data[0]:
img_tbl = Table(img_row_data, colWidths=[8.5*cm, 8.5*cm])
img_tbl.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),
("VALIGN",(0,0),(-1,-1),"MIDDLE"),
("LEFTPADDING",(0,0),(-1,-1),0),
("RIGHTPADDING",(0,0),(-1,-1),0)]))
cap_tbl = Table(img_row_captions, colWidths=[8.5*cm, 8.5*cm])
cap_tbl.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),
("TOPPADDING",(0,0),(-1,-1),2)]))
story += [img_tbl, cap_tbl, Spacer(1, 0.3*cm)]
# Key features box
features_loose = [
["Feature", "Details"],
["Fibers", "Collagen (type I), elastic, reticular — all present, randomly arranged"],
["Dominant Cell", "Fibroblasts; also macrophages, mast cells, plasma cells, leukocytes"],
["Ground Substance", "Abundant; hydrophilic; allows metabolic exchange"],
["Vascularity", "Richly vascularized; supports nearby epithelia"],
["Stain on H&E", "Pale pink; few dense structures; many nuclei of various shapes"],
["Locations", "Lamina propria, dermis, mesenteries, perivascular tissue, adventitia"],
]
story.append(_mini_table(features_loose))
story.append(Spacer(1, 0.5*cm))
# ── 2b Dense Irregular CT ────────────────────────────────────────────────
story.append(Paragraph("2b. Dense Irregular Connective Tissue", h2_style))
story.append(Paragraph(
"Dense irregular CT has the same components as loose CT but collagen greatly predominates. "
"Thick bundles of type I collagen are arranged randomly in multiple directions, providing "
"resistance to forces from any direction. Cells are sparse (almost exclusively fibroblasts). "
"Ground substance is minimal.",
body_style))
di_path = img_paths.get("dense_irregular")
ld_path = img_paths.get("loose_dense_histo")
img_row2 = [[]]
cap_row2 = [[]]
if ld_path and os.path.exists(ld_path):
img_row2[0].append(Image(ld_path, width=8*cm, height=5.5*cm))
cap_row2[0].append(Paragraph(
"Mallory-Azan: LCT (loose) above, DCT (dense)\nirregular below. Note sparser nuclei\nand denser collagen in DCT (Ross Histology)",
caption_style))
if di_path and os.path.exists(di_path):
img_row2[0].append(Image(di_path, width=8*cm, height=5.5*cm))
cap_row2[0].append(Paragraph(
"H&E: Dense irregular CT — randomly oriented\nthick collagen bundles, minimal cells\n(Junqueira's 17e)",
caption_style))
if img_row2[0]:
t1 = Table(img_row2, colWidths=[8.5*cm, 8.5*cm])
t1.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),("VALIGN",(0,0),(-1,-1),"MIDDLE"),
("LEFTPADDING",(0,0),(-1,-1),0),("RIGHTPADDING",(0,0),(-1,-1),0)]))
t2 = Table(cap_row2, colWidths=[8.5*cm, 8.5*cm])
t2.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),("TOPPADDING",(0,0),(-1,-1),2)]))
story += [t1, t2, Spacer(1, 0.3*cm)]
features_di = [
["Feature", "Details"],
["Fibers", "Thick type I collagen bundles, multi-directional; few elastic fibers"],
["Dominant Cell", "Fibroblasts only (sparse)"],
["Ground Substance", "Minimal"],
["Stain on H&E", "Densely pink; few, widely spaced nuclei"],
["Locations", "Dermis, organ capsules (kidney, testis), submucosa, periosteum, perichondrium"],
]
story.append(_mini_table(features_di))
story.append(Spacer(1, 0.5*cm))
# ── 2c Dense Regular CT ──────────────────────────────────────────────────
story.append(Paragraph("2c. Dense Regular Connective Tissue (Tendon)", h2_style))
story.append(Paragraph(
"Dense regular CT has parallel, tightly packed type I collagen bundles with fibroblasts "
"(tenocytes/tendinocytes) aligned in rows between them. It provides maximum tensile strength "
"along the axis of force. The tissue is glistening white macroscopically and largely avascular "
"(slow healing after injury).",
body_style))
ten_path = img_paths.get("tendon_longitudinal")
if ten_path and os.path.exists(ten_path):
story.append(Image(ten_path, width=13*cm, height=5*cm))
story.append(Paragraph(
"H&E ×100: Dense regular CT (tendon, longitudinal section). Homogeneous pink collagen fascicles (TF) with "
"tenocyte nuclei in single-file rows. Outer epitendineum (dense irregular CT) is visible at top.\n(Ross Histology 9e)",
caption_style))
story.append(Spacer(1, 0.3*cm))
features_dr = [
["Feature", "Details"],
["Fibers", "Parallel bundles of type I collagen; minimal elastic fibers"],
["Dominant Cell", "Tenocytes/tendinocytes (fibroblasts) in linear rows between bundles"],
["Ground Substance", "Minimal"],
["Stain on H&E", "Homogeneous pink; elongate nuclei in rows"],
["Vascularity", "Poorly vascularized → slow healing"],
["Locations", "Tendons, ligaments, aponeuroses, corneal stroma"],
]
story.append(_mini_table(features_dr))
story.append(PageBreak())
# ── 2d Reticular CT ──────────────────────────────────────────────────────
story.append(section_header("2. Histology (continued)", bg="#f5f7fa"))
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("2d. Reticular Connective Tissue", h2_style))
story.append(Paragraph(
"Reticular CT consists of a delicate, 3-dimensional network of type III collagen fibers "
"(reticulin) produced by specialized fibroblasts called reticular cells. The fibers are "
"heavily glycosylated, making them argyrophilic (stain black with silver salts). They support "
"rapidly changing populations of hematopoietic and immune cells.",
body_style))
ret1 = img_paths.get("reticular_fibers_1")
ret2 = img_paths.get("reticular_fibers_2")
img_row3 = [[]]
cap_row3 = [[]]
if ret1 and os.path.exists(ret1):
img_row3[0].append(Image(ret1, width=7.5*cm, height=6*cm))
cap_row3[0].append(Paragraph("Silver stain: Reticular fibers (black)\nin adrenal cortex ×100\n(Junqueira's 17e)", caption_style))
if ret2 and os.path.exists(ret2):
img_row3[0].append(Image(ret2, width=7.5*cm, height=6*cm))
cap_row3[0].append(Paragraph("Silver stain: Reticular network\nin lymph node ×100\n(Junqueira's 17e)", caption_style))
if img_row3[0]:
t1 = Table(img_row3, colWidths=[8.5*cm, 8.5*cm])
t1.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),("VALIGN",(0,0),(-1,-1),"MIDDLE"),
("LEFTPADDING",(0,0),(-1,-1),0),("RIGHTPADDING",(0,0),(-1,-1),0)]))
t2 = Table(cap_row3, colWidths=[8.5*cm, 8.5*cm])
t2.setStyle(TableStyle([("ALIGN",(0,0),(-1,-1),"CENTER"),("TOPPADDING",(0,0),(-1,-1),2)]))
story += [t1, t2, Spacer(1, 0.3*cm)]
features_ret = [
["Feature", "Details"],
["Fiber Type", "Type III collagen (reticulin) — thin, delicate, branching network"],
["Staining", "Argyrophilic (black with silver); PAS-positive; poorly visible on H&E"],
["Dominant Cell", "Reticular cells (specialized fibroblasts)"],
["Function", "Stroma for hematopoietic cells, lymphocytes, and secretory cells"],
["Carbohydrate Content", "~10% (vs 1% in type I collagen) — responsible for argyrophilia"],
["Locations", "Bone marrow, liver (sinusoids), spleen, all lymphoid organs, endocrine glands"],
]
story.append(_mini_table(features_ret))
story.append(Spacer(1, 0.5*cm))
# ── 2e Mesenchyme ─────────────────────────────────────────────────────────
story.append(Paragraph("2e. Embryonic Connective Tissue — Mesenchyme", h2_style))
story.append(Paragraph(
"Mesenchyme is the precursor of all adult connective tissues. It contains "
"undifferentiated, multipotent cells with large euchromatic nuclei and prominent nucleoli, "
"embedded in a sparse matrix rich in hyaluronan with very little collagen.",
body_style))
mes_path = img_paths.get("mesenchyme")
if mes_path and os.path.exists(mes_path):
story.append(Image(mes_path, width=11*cm, height=6*cm))
story.append(Paragraph(
"Mallory trichrome ×200: Embryonic mesenchyme — undifferentiated elongated cells\n"
"with large pale nuclei; sparse matrix with minimal collagen. (Junqueira's 17e)",
caption_style))
story.append(Spacer(1, 0.6*cm))
# ── SECTION 3 – FIBERS ────────────────────────────────────────────────────
story.append(PageBreak())
story.append(section_header("3. Connective Tissue Fibers"))
story.append(Spacer(1, 0.3*cm))
# Fiber comparison table
fiber_data = [
["Property", "Collagen (Type I)", "Reticular (Type III)", "Elastic Fibers"],
["Protein", "Collagen type I", "Collagen type III", "Elastin + fibrillin microfibrils"],
["Diameter", "1–20 μm (bundles)", "0.5–2 μm", "0.2–1 μm"],
["Structure", "Triple helix; parallel cross-linked fibrils", "Thin branching network (reticulum)", "Core of cross-linked elastin + fibrillin sheath"],
["Cross-links", "Lysyl oxidase (hydroxylysine aldehyde)", "Same; plus desmosine-like bonds", "Desmosine rings (from lysine); unique to elastin"],
["H&E stain", "Pink (eosinophilic)", "Poorly visible", "Pale, barely visible"],
["Special stain", "Masson trichrome (blue/green)", "Silver impregnation (black/argyrophilic)", "Orcein / Weigert's elastic stain (dark brown)"],
["PAS stain", "Weakly positive", "Strongly positive (~10% carbohydrate)", "Negative"],
["Tensile strength", "High — resists pulling forces", "Low — delicate support only", "Extensible (up to 150% length) then recoils"],
["Synthesized by", "Fibroblasts (+ osteoblasts, chondrocytes)", "Fibroblasts / reticular cells", "Fibroblasts + smooth muscle cells"],
["Key locations", "Tendons, dermis, bone, cornea", "Bone marrow, liver, lymph nodes", "Large arteries, lung, ligamentum flavum, skin"],
["Vitamin C\ndependence", "Yes — hydroxylation of Pro/Lys in RER", "Yes", "No"],
["Clinical disorders", "Scurvy (↓Vit C), Osteogenesis imperfecta\n(COL1A1/2 mutations), Ehlers-Danlos",
"Liver cirrhosis (↑ reticulin), fibrosis", "Marfan syndrome (FBN1), cutis laxa (elastin)"],
]
fcol = [3.5*cm, 4.3*cm, 4.3*cm, 4.4*cm]
fiber_tbl = Table(
[[Paragraph(f"<b>{c}</b>" if r == 0 else c,
make_style(f"ft{r}{i}", "Normal", fontSize=8, leading=11,
fontName="Helvetica-Bold" if r == 0 else "Helvetica",
textColor=colors.white if r == 0 else colors.black,
alignment=TA_CENTER if r == 0 else TA_LEFT))
for i, c in enumerate(row)]
for r, row in enumerate(fiber_data)],
colWidths=fcol, repeatRows=1
)
fiber_tbl.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#2c5f8a")),
("BACKGROUND", (1, 0), (1, 0), colors.HexColor("#8b2c2c")), # col headers color
("BACKGROUND", (0, 1), (0, -1), colors.HexColor("#f0f4fa")), # property column
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f7f9fc") if i % 2 == 0 else colors.white)
for i in range(len(fiber_data)-1)],
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 4),
("BOTTOMPADDING",(0, 0), (-1, -1), 4),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTNAME", (0, 0), (-1, 0), "Helvetica-Bold"),
("FONTSIZE", (0, 0), (-1, 0), 8.5),
("ALIGN", (0, 0), (-1, 0), "CENTER"),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
]))
story.append(fiber_tbl)
story.append(Spacer(1, 0.4*cm))
# Elastic fibers image
el_path = img_paths.get("elastic_fibers")
if el_path and os.path.exists(el_path):
story.append(Image(el_path, width=14*cm, height=5*cm))
story.append(Paragraph(
"Elastic fibers in connective tissue. Left: H&E (fibers barely visible). "
"Center/Right: orcein or aldehyde fuchsin stain highlights elastic fibers as dark threads. "
"(Junqueira's Basic Histology 17e)",
caption_style))
story.append(PageBreak())
# ── SECTION 4 – CELLS TABLE ───────────────────────────────────────────────
story.append(section_header("4. Cells of Connective Tissue"))
story.append(Spacer(1, 0.3*cm))
cell_data = [
["Cell", "Origin", "Morphology", "Key Functions", "Special Features"],
["Fibroblast /\nFibrocyte", "Mesenchyme\n(permanent resident)", "Spindle-shaped; pale euchromatic nucleus; abundant RER (active form)",
"Synthesize ALL ECM: collagen, elastin, GAGs, proteoglycans, glycoproteins",
"Fibrocyte = inactive form; activated in wound healing"],
["Adipocyte\n(White)", "Mesenchyme", "Large; single lipid droplet; peripherally displaced nucleus; 'signet ring'",
"Energy storage; thermal insulation; secretes leptin, adiponectin",
"Cytoplasm and lipid clear on H&E"],
["Adipocyte\n(Brown)", "Mesenchyme", "Smaller; multiple lipid droplets; central nucleus; mitochondria-rich",
"Non-shivering thermogenesis (UCP-1 uncouples oxidative phosphorylation)",
"Abundant in neonates; diminishes with age"],
["Mast Cell", "Bone marrow\nprogenitors", "Ovoid; metachromatic granules (purple/toluidine blue); bilobed nucleus",
"Release histamine, heparin, cytokines, leukotrienes; immediate hypersensitivity; anti-parasite",
"IgE receptors on surface; perivascular and mucosal locations; sentinel cells"],
["Macrophage\n(Histiocyte)", "Bone marrow monocyte", "Irregular; kidney-shaped nucleus; pseudopods; many lysosomes",
"Phagocytosis; antigen presentation (MHC II); secrete cytokines (TNF-α, IL-1, IL-6)",
"Specialized forms: Kupffer (liver), microglia (brain), osteoclasts (bone)"],
["Plasma Cell", "B lymphocyte", "Large ovoid; basophilic (RER-rich); 'clock-face' nucleus; perinuclear halo (Golgi)",
"Secrete immunoglobulins (antibodies)",
"Abundant in GI lamina propria and mucosal sites"],
["Lymphocyte", "Bone marrow", "Small; large dark nucleus; scant cytoplasm",
"Adaptive immunity: T cells (cellular), B cells (humoral), NK cells",
"Normally sparse; increase dramatically at infection sites"],
["Neutrophil", "Bone marrow", "Multilobed nucleus (3–5 lobes); granules contain lysozyme, myeloperoxidase",
"First responder; phagocytosis of bacteria; acute inflammation",
"Short-lived (~days); most abundant WBC"],
["Eosinophil", "Bone marrow", "Bilobed nucleus; large eosinophilic (pink) cytoplasmic granules",
"Defense against parasites; modulate IgE/allergic reactions; phagocytose immune complexes",
"Major basic protein in granules is toxic to parasites"],
]
ccol = [2.5*cm, 2.8*cm, 3.5*cm, 4.2*cm, 4.0*cm]
cell_tbl = Table(
[[Paragraph(f"<b>{c}</b>" if r == 0 else c,
make_style(f"ct{r}{i}", "Normal", fontSize=7.8, leading=11,
fontName="Helvetica-Bold" if r == 0 else "Helvetica",
textColor=colors.white if r == 0 else colors.black))
for i, c in enumerate(row)]
for r, row in enumerate(cell_data)],
colWidths=ccol, repeatRows=1
)
cell_tbl.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#1a3d6b")),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
("FONTNAME", (0, 0), (-1, 0), "Helvetica-Bold"),
("ALIGN", (0, 0), (-1, 0), "CENTER"),
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(cell_data)-1)],
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 4),
("BOTTOMPADDING",(0, 0), (-1, -1), 4),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
]))
story.append(cell_tbl)
story.append(PageBreak())
# ── SECTION 5 – ECM / GROUND SUBSTANCE ───────────────────────────────────
story.append(section_header("5. Extracellular Matrix (ECM)"))
story.append(Spacer(1, 0.3*cm))
# ECM overview image
ecm_path = img_paths.get("ecm_overview")
if ecm_path and os.path.exists(ecm_path):
story.append(Image(ecm_path, width=13*cm, height=7*cm))
story.append(Paragraph(
"ECM structure: Basal lamina beneath the epithelial cell layer, with collagen fibers, "
"elastic fibers, and proteoglycans forming the interstitial matrix. (Basic Medical Biochemistry 6e)",
caption_style))
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("5a. Glycosaminoglycans (GAGs)", h3_style))
story.append(Paragraph(
"Long, unbranched polysaccharide chains of repeating disaccharide units (hexosamine + uronic acid). "
"Highly negatively charged (sulfated except hyaluronan) → attract water → gel-like ground substance. "
"Degraded by lysosomal enzymes; deficiency causes <b>mucopolysaccharidoses</b> (e.g., Hurler, Hunter syndromes).",
body_style))
gag_data = [
["GAG", "Sulfated?", "Key Features", "Major Locations"],
["Hyaluronan", "No", "Largest GAG; not covalently linked to protein; extremely hydrophilic; backbone of proteoglycan aggregates",
"Ubiquitous: synovial fluid, vitreous, umbilical cord, embryonic tissues"],
["Chondroitin sulfate", "Yes", "Most abundant GAG in body; linked to core proteins (aggrecan)",
"Cartilage, bone, tendon, skin, cornea"],
["Dermatan sulfate", "Yes", "Associated with type I collagen; modulates fibrillogenesis",
"Skin, blood vessel walls, heart valves, tendons"],
["Heparan sulfate", "Yes", "Component of basal laminae; cell surface proteoglycans (syndecan)",
"Basement membranes, cell surfaces"],
["Heparin", "Highly\nsulfated", "Stored in mast cell granules; anticoagulant",
"Mast cells; used clinically as anticoagulant"],
["Keratan sulfate", "Yes", "Lacks uronic acid; linked to protein via Asn or Ser",
"Cornea (type I), cartilage (type II), nucleus pulposus"],
]
gcol = [3*cm, 2*cm, 6.5*cm, 5.5*cm]
gag_tbl = Table(
[[Paragraph(f"<b>{c}</b>" if r == 0 else c,
make_style(f"gg{r}{i}", "Normal", fontSize=8, leading=11,
fontName="Helvetica-Bold" if r == 0 else "Helvetica",
textColor=colors.white if r == 0 else colors.black))
for i, c in enumerate(row)]
for r, row in enumerate(gag_data)],
colWidths=gcol, repeatRows=1
)
gag_tbl.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#2c5f8a")),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
("FONTNAME", (0, 0), (-1, 0), "Helvetica-Bold"),
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(gag_data)-1)],
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 3),
("BOTTOMPADDING",(0, 0), (-1, -1), 3),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
]))
story.append(gag_tbl)
story.append(Spacer(1, 0.4*cm))
story.append(Paragraph("5b. Multiadhesive Glycoproteins", h3_style))
glyco_data = [
["Glycoprotein", "Binds To", "Function"],
["Fibronectin", "Collagen, heparan sulfate, integrins, fibrin",
"Cell adhesion & migration; wound healing; matrix assembly; fibroblast movement"],
["Laminin", "Type IV collagen, nidogen, perlecan, integrins",
"Major component of basal laminae; cell attachment, differentiation, polarization"],
["Nidogen (Entactin)", "Laminin + type IV collagen",
"Bridges laminin and type IV collagen in basal lamina"],
["Tenascin", "Fibronectin, proteoglycans",
"Anti-adhesive; modulates cell-matrix interactions; wound healing and tumor stroma"],
]
gcol2 = [3.5*cm, 5.5*cm, 8*cm]
glyco_tbl = _data_table(glyco_data, gcol2)
story.append(glyco_tbl)
story.append(Spacer(1, 0.4*cm))
story.append(Paragraph("5c. Integrins and MMPs", h3_style))
story.append(Paragraph(
"<b>Integrins</b>: Transmembrane heterodimeric receptors (α+β subunits) that bind ECM components "
"(fibronectin, laminin, collagen) extracellularly and link to the actin cytoskeleton intracellularly "
"(via talin, vinculin). They transduce bi-directional signals between the ECM and cell interior, "
"regulating migration, differentiation, proliferation, and survival.",
body_style))
story.append(Paragraph(
"<b>MMPs (Matrix Metalloproteinases)</b>: Zinc-dependent endopeptidases that degrade ECM components. "
"Regulated by <b>TIMPs</b> (tissue inhibitors of MMPs). Dysregulation promotes cancer invasion, "
"metastasis, atherosclerotic plaque rupture, and fibrosis.",
body_style))
story.append(PageBreak())
# ── SECTION 6 – QUICK REFERENCE ──────────────────────────────────────────
story.append(section_header("6. Quick Reference – Locations & Special Stains"))
story.append(Spacer(1, 0.3*cm))
loc_data = [
["Location / Organ", "Primary CT Type", "Predominant Fiber", "Key Cells"],
["GI tract lamina propria", "Loose (areolar)", "Collagen I + reticular", "Plasma cells, lymphocytes, macrophages, mast cells"],
["Skin dermis (upper)", "Loose → Dense irregular", "Collagen I (+ elastic)", "Fibroblasts, macrophages, mast cells"],
["Skin dermis (deep)", "Dense irregular", "Collagen I (thick bundles)", "Fibroblasts"],
["Tendons", "Dense regular", "Collagen I (parallel)", "Tenocytes"],
["Ligaments", "Dense regular", "Collagen I (parallel, + elastic in ligamentum flavum)", "Fibroblasts"],
["Bone marrow stroma", "Reticular", "Collagen III (reticulin)", "Reticular cells, hematopoietic cells"],
["Liver sinusoids (space of Disse)", "Reticular", "Collagen III (reticulin)", "Reticular cells, Kupffer cells"],
["Lymph nodes / Spleen", "Reticular", "Collagen III", "Reticular cells, lymphocytes, macrophages"],
["Large artery wall (tunica media)", "Elastic (smooth muscle + elastic lamellae)", "Elastin lamellae + collagen I", "Smooth muscle cells"],
["Organ capsules (kidney, testis)", "Dense irregular", "Collagen I", "Fibroblasts"],
["Articular cartilage", "Hyaline cartilage", "Collagen II", "Chondrocytes"],
["Intervertebral disc (annulus)", "Fibrocartilage", "Collagen I (predominant)", "Fibrocartilage chondrocytes"],
["Cornea", "Dense regular (orthogonal layers)", "Collagen I (+ V)", "Keratocytes (fibroblasts)"],
["Umbilical cord", "Mucoid (Wharton's jelly)", "Sparse collagen + hyaluronan", "Fibroblasts"],
["Subcutaneous tissue", "Loose + White adipose", "Collagen I + reticular", "Adipocytes, fibroblasts"],
["Lung alveolar walls", "Loose", "Elastic + reticular + collagen III", "Fibroblasts, macrophages"],
["Mesentery", "Loose", "Collagen I, elastic, reticular", "All CT cell types"],
]
lcol = [4.5*cm, 3.5*cm, 3.8*cm, 5.2*cm]
loc_tbl = Table(
[[Paragraph(f"<b>{c}</b>" if r == 0 else c,
make_style(f"lc{r}{i}", "Normal", fontSize=8, leading=11,
fontName="Helvetica-Bold" if r == 0 else "Helvetica",
textColor=colors.white if r == 0 else colors.black))
for i, c in enumerate(row)]
for r, row in enumerate(loc_data)],
colWidths=lcol, repeatRows=1
)
loc_tbl.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#1a3d6b")),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
("FONTNAME", (0, 0), (-1, 0), "Helvetica-Bold"),
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(loc_data)-1)],
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 3),
("BOTTOMPADDING",(0, 0), (-1, -1), 3),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
("FONTSIZE", (0, 1), (0, -1), 8),
]))
story.append(loc_tbl)
story.append(Spacer(1, 0.4*cm))
# Special stains table
story.append(Paragraph("Histological Staining Reference", h3_style))
stain_data = [
["Stain", "What It Colors", "CT Component Highlighted"],
["H&E (Hematoxylin & Eosin)", "Nuclei: blue/purple; cytoplasm/collagen: pink", "General morphology; collagen appears eosinophilic"],
["Masson's Trichrome", "Collagen: blue or green; muscle: red; nuclei: dark", "Collagen fibers (all types visible)"],
["Gomori / Reticulin (silver)", "Reticular fibers: black; other tissue: pale", "Type III collagen (reticulin) specifically"],
["Orcein / Aldehyde Fuchsin", "Elastic fibers: dark brown/black", "Elastic fibers and lamellae"],
["Weigert's Elastic Stain", "Elastic fibers: blue-black", "Elastic fibers"],
["PAS (Periodic acid-Schiff)", "Polysaccharides, glycoproteins: magenta", "Basement membranes; reticular fibers (high CHO content)"],
["Toluidine Blue", "GAGs: metachromatic (purple/red)", "Mast cell granules; cartilage matrix; heparin"],
["Alcian Blue", "Acid GAGs: blue", "Cartilage proteoglycans; mucins"],
["Sudan IV / Oil Red O", "Neutral lipids: red", "Adipocytes; lipid droplets"],
["Von Kossa", "Calcium phosphate: black/brown", "Calcified bone; cartilage ossification"],
]
scol = [4.5*cm, 5.5*cm, 7*cm]
story.append(_data_table(stain_data, scol))
story.append(Spacer(1, 0.4*cm))
# ── FOOTER / REFERENCES ───────────────────────────────────────────────────
story.append(HRFlowable(width=W, thickness=1, color=colors.HexColor("#1a3d6b")))
story.append(Spacer(1, 0.2*cm))
ref_style = make_style("Ref","Normal", fontSize=8, leading=13,
textColor=colors.HexColor("#333333"))
story.append(Paragraph("<b>References</b>", ref_style))
story.append(Paragraph(
"1. Mescher AL. <i>Junqueira's Basic Histology: Text and Atlas</i>, 17th ed. McGraw Hill, 2024. (ISBN 9781264930395)\n"
"2. Ross MH, Pawlina W. <i>Histology: A Text and Atlas with Correlated Cell and Molecular Biology</i>, 9th ed. Wolters Kluwer, 2021. (ISBN 9781975181512)\n"
"3. Lieberman M, Peet A. <i>Basic Medical Biochemistry: A Clinical Approach</i>, 6th ed. LWW, 2022. (ISBN 9781975150143)",
make_style("refs2","Normal",fontSize=8,leading=13,textColor=colors.HexColor("#444444"))))
doc.build(story)
print(f"\n✅ PDF created: {OUTPUT}")
# ── Helper tables ──────────────────────────────────────────────────────────────
def _mini_table(data):
col_w = [4.5*cm, 12.5*cm]
rows = []
for i, row in enumerate(data):
bg = colors.HexColor("#1a3d6b") if i == 0 else (
colors.HexColor("#f0f4fa") if i % 2 == 1 else colors.white)
fc = colors.white if i == 0 else colors.black
fn = "Helvetica-Bold" if i == 0 else "Helvetica"
rows.append([
Paragraph(row[0], make_style(f"mt{i}a","Normal",fontSize=8.5,
fontName="Helvetica-Bold", textColor=fc)),
Paragraph(row[1], make_style(f"mt{i}b","Normal",fontSize=8.5,
textColor=fc, fontName=fn)),
])
t = Table(rows, colWidths=col_w)
t.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#1a3d6b")),
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(data)-1)],
("GRID", (0, 0), (-1, -1), 0.3, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 3),
("BOTTOMPADDING",(0, 0), (-1, -1), 3),
("LEFTPADDING", (0, 0), (-1, -1), 5),
("RIGHTPADDING", (0, 0), (-1, -1), 5),
("VALIGN", (0, 0), (-1, -1), "MIDDLE"),
]))
return t
def _data_table(data, col_widths):
rows = []
for i, row in enumerate(data):
rows.append([
Paragraph(f"<b>{c}</b>" if i == 0 else c,
make_style(f"dt{i}{j}","Normal", fontSize=8, leading=11,
fontName="Helvetica-Bold" if i == 0 else "Helvetica",
textColor=colors.white if i == 0 else colors.black))
for j, c in enumerate(row)
])
t = Table(rows, colWidths=col_widths, repeatRows=1)
t.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), colors.HexColor("#2c5f8a")),
("TEXTCOLOR", (0, 0), (-1, 0), colors.white),
*[("BACKGROUND", (0, i+1), (-1, i+1),
colors.HexColor("#f0f4fa") if i % 2 == 0 else colors.white)
for i in range(len(data)-1)],
("GRID", (0, 0), (-1, -1), 0.4, colors.HexColor("#c0cce0")),
("TOPPADDING", (0, 0), (-1, -1), 3),
("BOTTOMPADDING",(0, 0), (-1, -1), 3),
("LEFTPADDING", (0, 0), (-1, -1), 4),
("RIGHTPADDING", (0, 0), (-1, -1), 4),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTNAME", (0, 1), (0, -1), "Helvetica-Bold"),
]))
return t
if __name__ == "__main__":
build()
cd /home/daytona/workspace/connective-tissue-pdf && python3 build_pdf.py 2>&1
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
~/connective-tissue-pdf/build_pdf.py
Successfully replaced 1 instance(s) of the string in '/home/daytona/workspace/connective-tissue-pdf/build_pdf.py'
cd /home/daytona/workspace/connective-tissue-pdf && python3 build_pdf.py 2>&1 && echo "SUCCESS" && ls -lh Connective_Tissue_Summary.pdf