Acute paronychia.
Reading File
Finding Sources
Searching PubMed
acute paronychia treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative textbooks. Here is a detailed, well-organized answer.
Acute Paronychia
Definition
Acute paronychia is a bacterial infection of the soft tissue fold (nail fold) surrounding the nail root. It is one of the most common infections of the hand. The term comes from the Greek "para" (beside) + "onyx" (nail) and is sometimes called a "runaround" infection. - Campbell's Operative Orthopaedics 15e
Anatomy
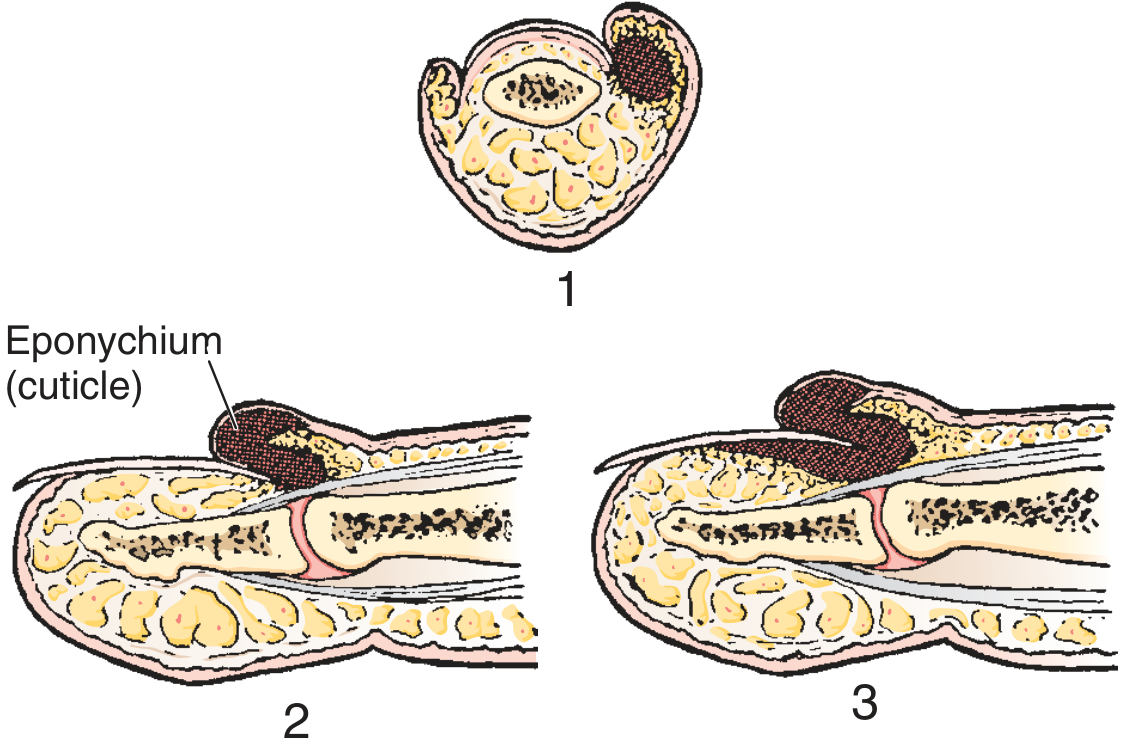
The infection involves the eponychium (cuticle/proximal nail fold) and the paronychium (lateral nail folds). Pus collects in the potential space between the cuticle and the proximal end of the fingernail. The infection begins superficially but can spread under the nail to form a subungual abscess.
Aetiology / Causative Organisms
- Most common: Staphylococcus aureus (including CA-MRSA)
- Streptococci (including Group A beta-haemolytic streptococci - especially in thumb-sucking children with streptococcal pharyngitis)
- Anaerobes (especially in children; associated with finger-sucking and nail-biting)
- Mixed aerobic + anaerobic - 50% of cases in one series; 25% pure anaerobic, 25% pure aerobic
- Gram-negative bacilli (e.g. Pseudomonas aeruginosa, E. coli) - especially in patients with oral flora exposure
- Non-bacterial: viruses (Herpes simplex - herpetic whitlow; must be excluded) and yeast
Predisposing factors:
-
Hangnails, nail-biting, finger-sucking
-
Nail cosmetics (mechanical trauma, irritant/allergic reactions)
-
Occupations: hairdressing, meat-handling
-
HIV/antiretroviral therapy
-
Epidermal growth factor receptor inhibitors
-
Roberts & Hedges' Clinical Procedures in Emergency Medicine; Campbell's Operative Orthopaedics 15e
Clinical Features
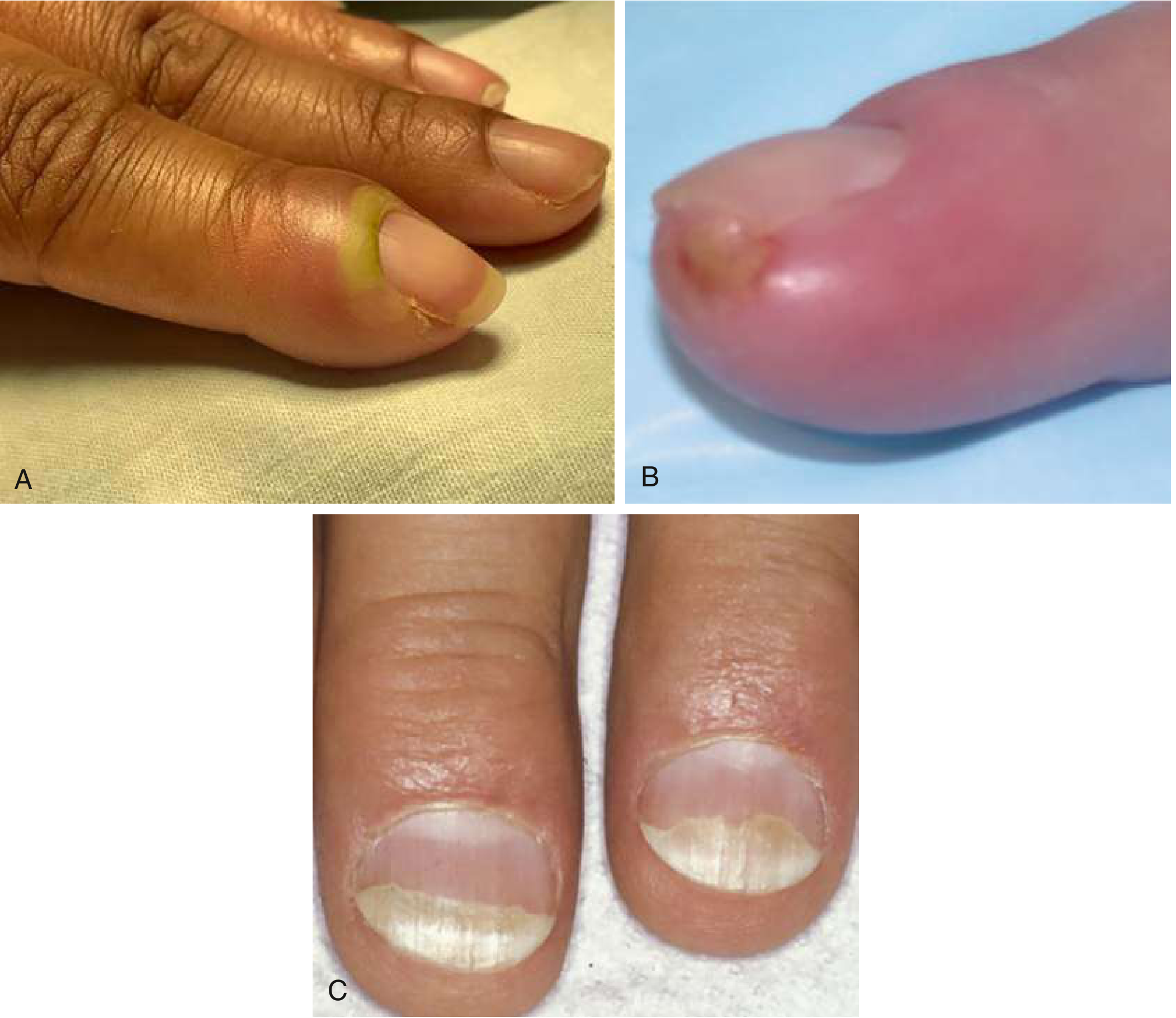
Symptoms and signs:
- Pain (often around a hangnail) - the usual presenting complaint
- Redness, swelling, tenderness of the soft tissue along the base or side of the nail
- Fluctuance when abscess forms
- Pus visible beneath or around the nail fold
Progression (three stages):
- Abscess at the side of the nail (lateral fold only)
- Infection spreads around the base of the nail, raising the eponychium (but not yet under the nail)
- End stage - subeponychial and subungual abscess (nail becomes mobile, loses normal lustre)
Key sign: If the nail bed is mobile, the infection has extended under the nail and a more extensive drainage procedure is required. - Roberts & Hedges
Management
Management depends entirely on the stage of infection.
1. Early (Cellulitic) Stage - No Abscess
- Warm soaks: 3x/day (warm water, or warm water + 1% acetic acid for 15 min, 2-4x/day)
- Topical antibiotics: mupirocin daily for 7-10 days ± topical steroid at night to reduce inflammation
- Oral antibiotics if significant cellulitis is present: broad-spectrum antistaphylococcal agent
Incision has little value in the early cellulitic phase. - Roberts & Hedges
2. Abscess Stage - Definitive: Incision and Drainage (I&D)
Drainage technique:
- Perform a digital nerve block first
- Soften eponychium by soaking
- Advance a No. 11 scalpel blade, scissors, or a 21-23 gauge needle parallel to the nail and under the eponychium at the site of maximal swelling
- Simply lift the eponychial fold away from the nail matrix - this allows pus to drain and is usually curative
- No cutting of skin is required in most cases; no drain is needed
- Do NOT use skin incision or nail removal as the initial treatment
If abscess is under one corner of the nail root:
- Remove that corner of nail
If abscess has migrated to the opposite side or under the nail:
- Make a second incision, fold skin proximally, excise the proximal one-third of the nail
- Pack loosely with iodoform gauze for 48 hours
- Trim any loose/dead nail fragments carefully
Nail resection:
- Required only if the nail base is undermined by pus
- The loose nail will lack its normal lustre and should be removed to allow free drainage
"Pus is released by stripping back the cuticle and lateral nail folds from the surface of the nail, using the closed points of fine dissecting forceps. No cutting is needed, except to trim away any loose portions of cuticle." - Pye's Surgical Handicraft 22e
3. Antibiotics Post-Drainage
With adequate I&D, post-procedural antibiotics are often unnecessary in immunocompetent patients. Prescribe judiciously based on comorbidities and clinical judgment. - Campbell's Operative Orthopaedics 15e; Rosen's Emergency Medicine
Antibiotic choices (when indicated):
| Organism(s) Suspected | Agent |
|---|---|
| S. aureus / Streptococci | First-generation cephalosporin (e.g. cefalexin) OR antistaphylococcal penicillin |
| Anaerobes / oral flora | Clindamycin OR amoxicillin-clavulanate (co-amoxiclav) |
| CA-MRSA | Trimethoprim-sulfamethoxazole or clindamycin |
| Pseudomonas (water exposure) | Soaks with 1% acetic acid; topical agents |
- Roberts & Hedges Table 57.1
Complications
- Subungual abscess - requires partial or complete nail removal
- Felon - spread to the finger pulp (a distinct and serious separate infection)
- Flexor tenosynovitis - rare but severe
- Osteomyelitis of the distal phalanx - rare
- Inadequate resection leads to recurrence - Campbell's Operative Orthopaedics
Differential Diagnosis
- Herpetic whitlow (HSV infection) - vesicular, not pustular; I&D is contraindicated; treat with antivirals if <48 hours onset
- Chronic paronychia - duration >6 weeks; usually Candida or irritant/hypersensitivity; keep dry, topical antifungal (terbinafine 1% cream BD) ± topical steroid
- Felon (pulp space infection)
- Ingrown toenail (onychocryptosis)
Summary
| Stage | Finding | Treatment |
|---|---|---|
| Early cellulitis | Redness, swelling, no pus | Warm soaks + oral antibiotics |
| Frank abscess | Fluctuance, pus | Digital block + I&D (lift eponychium) ± antibiotics |
| Subungual extension | Mobile nail, pus under nail | Partial/complete nail removal + I&D |
| Bilateral/extensive | Pus on both sides, under base | Double incision + proximal 1/3 nail excision |
Sources: Roberts & Hedges' Clinical Procedures in Emergency Medicine; Rosen's Emergency Medicine; Campbell's Operative Orthopaedics 15e (2026); Pye's Surgical Handicraft 22e; Bailey & Love's Short Practice of Surgery 28e
In-growing toe nail.
Finding Sources
Searching PubMed
ingrown toenail onychocryptosis treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content covering all aspects. Let me compile the full answer.
Ingrown Toenail (Onychocryptosis / Unguis Incarnatus)
Definition
An ingrown toenail is a condition in which the lateral margin of the nail plate penetrates or impinges upon the adjacent periungual soft tissue (nail fold), causing pain, inflammation, and eventually infection and granulation tissue formation. It occurs predominantly on the great toe (hallux). - Roberts & Hedges' Clinical Procedures in Emergency Medicine
Pathology
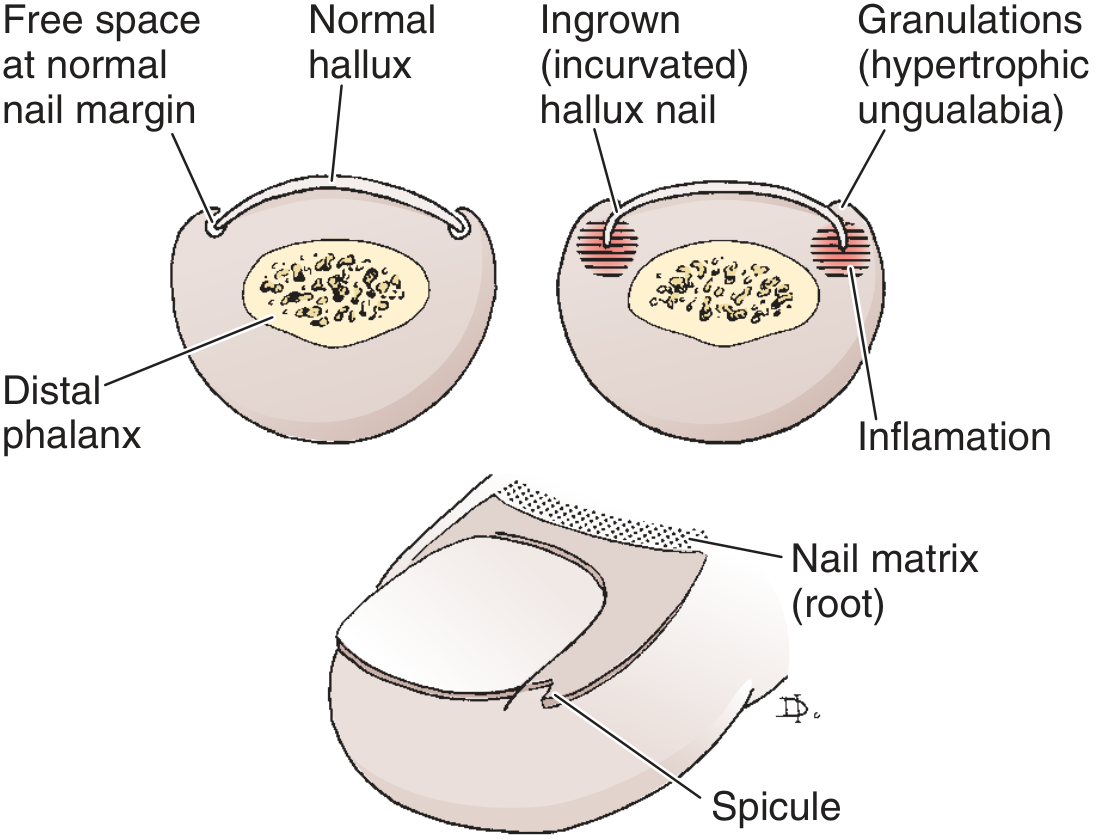
The normal toenail grows distally with free space at the lateral nail margin. In onychocryptosis, the lateral edge of the nail acts as a foreign body, breaching the skin. Skin bacteria and fungi enter the wound, causing:
- Inflammation → erythema, oedema, hyperhidrosis, tenderness
- Bottlenecked poorly draining abscess
- Hypertrophic granulation tissue (ungualabia) that covers the nail fold, further inhibiting drainage
- Epithelium creeps over granulations → perpetuates the cycle
The condition is rare in people who do not wear shoes, indicating extrinsic pressure (shoe toe box) is central to the pathogenesis. - Campbell's Operative Orthopaedics 15e
Aetiology / Predisposing Factors
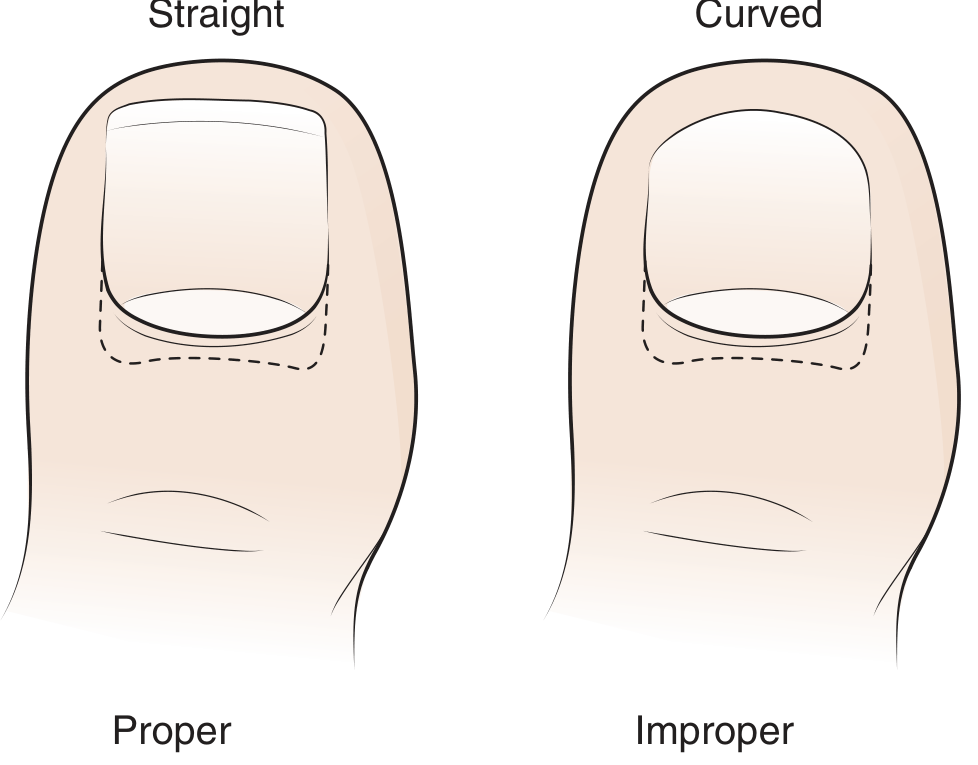
- Improper nail trimming - cutting the nail in a curve rather than straight across (most common cause)
- Ill-fitting footwear - lateral pressure from shoe toe box pushes the great toe against the second toe
- Nail deformity (incurvated/pincer nail)
- Rotational deformity of the toes
- Tight hosiery
- Trauma to the nail matrix
- Drugs: targeted cancer therapies, isotretinoin, lamivudine, indinavir (cause periungual granulation tissue mimicking onychocryptosis)
Clinical Stages (Heifetz / Park Classification)
| Stage | Features | Treatment |
|---|---|---|
| Stage I (Inflammatory) | Mild erythema, swelling, tenderness of lateral nail fold; no abscess | Conservative: soaks, cotton elevation, nail splinting |
| Stage II (Abscess) | Stage I + bulging nail fold, drainage (initially serous → purulent), fetid odour; wearing shoes almost impossible | Usually operative; can try conservative initially |
| Stage III (Granulation/Chronic) | Hypertrophic granulation tissue covering lateral nail fold; epithelium creeping over granulations; recurrent acute episodes | Operative - surgery preferred |
Clinical Features
- Pain - often severe; the hallmark
- Erythema and swelling of the lateral nail fold
- Fluctuance when abscess forms
- Purulent discharge (fetid odour in stage II)
- Hypertrophic granulation tissue in stage III
- Pressure over nail margin worsens pain
- Walking becomes difficult; wearing shoes almost impossible in advanced cases
Key evaluation point: When the free edge of the lateral nail can easily be visualized as separate from the lateral nail fold, consider other diagnoses - gout, trauma, paronychia, cellulitis. - Roberts & Hedges
Complications to consider: In patients with diabetes, peripheral neuropathy, or circulatory dysfunction, recurrent or neglected cases may develop osteomyelitis of the distal phalanx.
Differential Diagnosis
- Acute paronychia (bacterial infection of nail fold)
- Gout (first MTP is typical site)
- Cellulitis/soft tissue infection
- Felon (pulp space abscess)
- Subungual haematoma/trauma
- Verruca (plantar wart at nail margin)
- Onychomycosis (thickened, dystrophic nail)
Management
Anaesthesia
For any invasive procedure: digital nerve block (lidocaine or mepivacaine without adrenaline), introduced 1 cm distal to the first web space, ensuring plantar digital nerves and dorsal sensory branches of the superficial peroneal nerve are anaesthetised. A tourniquet (Penrose drain or gloved finger) ensures a bloodless field. - Campbell's Operative Orthopaedics 15e
Stage I - Conservative (Non-operative)
- Warm soaks - 10-15 min, 3-5 times/day to soften nail
- Cotton/wool elevation technique - lift the lateral nail edge from its embedded position; insert non-absorbent cotton, dental floss, or acrylic mesh beneath the corner of the nail; patient repeats daily until nail grows out and can be trimmed properly. Success in 2-3 weeks if done consistently.
- Proper nail trimming - straight across (not curved), with corners protruding just distal to the hyponychium
- Gutter splint technique - a sterilised vinyl IV drip tube slit lengthwise, inserted along the nail groove and affixed with cyanoacrylate or adhesive tape, providing a channel for the nail to grow free of the fold. Recurrence rates 8-48%.
- Orthonyxia (nail bracing) - two hooks on the sides of the nail connected under tension by a super-elastic wire; straightens the nail within ~3 weeks. Better cosmesis and shorter recovery than surgery.
- "Band-aid" method - adhesive tape pulls the nail fold away from the nail to relieve pressure.
Stage II - Operative (or Conservative if very early)
If attempting non-operative management in early stage II: remove all pressure from the toe, soak 4-5 times/day, culture drainage, start broad-spectrum antibiotics; cotton elevation once swelling and drainage resolve.
When an abscess is present or conservative management fails: surgical I&D and nail spicule removal.
Removal of the nail spicule (simple/ED procedure)
- Digital nerve block + tourniquet
- Separate nail from nail bed with scissors/elevator advanced parallel to nail bed
- Split nail lengthwise toward the cuticle with iris scissors
- Grasp the cut portion with a haemostat and twist toward the remaining nail to avulse
- Inspect for and remove any residual nail spicule (a retained spicule is the most common cause of recurrence)
- Debride inflamed/granulation tissue
Stage III - Definitive Surgical Treatment
Operative approaches aim to either remove the offending nail (nail-centric view) or debulk/remove the hypertrophied nail fold (soft tissue-centric view).
1. Total Nail Plate Removal (Avulsion) - Technique 89.42
Rarely the definitive treatment alone (nail regrows and may re-offend). Indicated when abscess has circumducted both sides. Technique:
- Pass thin hemostat beneath nail in midline from hyponychium to lunula
- Repeat at each lateral margin
- Extract with distal pull; use sharp dissection at eponychium if adherent
- Post-op: non-adherent dressing, compression bandage, elevate foot 24 h, then warm soaks; no constricting footwear for 1 week
- Nail re-forms in 4-6 months (patient must be warned of turned-up pulp deformity with repeated avulsions)
2. Partial Nail Plate Removal - Technique 89.43
- Remove only the lateral quarter to third of the nail plate
- Preferred in adolescents when cosmetic preservation of the nail is important
- High recurrence rate without matrix ablation
- Risk of nail spicule formation and permanent nail deformity (patient/parents must be counselled)
3. Partial Nail Plate Removal + Chemical Matricectomy (Phenolisation) - Technique 89.44 (Most common definitive procedure)
The most widely used curative procedure. Combines partial nail avulsion with ablation of the germinal matrix using 80-89% phenol:
Steps:
- All personnel wear gloves (phenol is corrosive)
- Tourniquet at base of great toe
- Elevate the lateral quarter to fifth of nail edge longitudinally, from distal to proximal (including nail beneath the eponychium)
- Apply antibiotic gel around the nail fold to protect surrounding skin
- Insert a phenol-soaked cotton swab into the nail groove, extending beneath the eponychium, for 4 minutes without interruption
- Wash the area with isopropyl alcohol to neutralise residual phenol
Alternative ablative agents: Trichloroacetic acid, 10% sodium hydroxide, silver nitrate (especially in adolescents), CO₂ laser, electrocoagulation, cryotherapy (liquid nitrogen, 20-30 sec freeze - reserved for those not suitable for other approaches)
Outcomes of phenolisation:
- Phenol matricectomy: recurrence 18-32%
- Surgical (excisional) matricectomy: recurrence 7-8% but higher infection, pain, and cosmetic dissatisfaction
- One RCT found both equally effective at 1 year
- Trichloroacetic acid/NaOH: similar efficacy to phenol; faster healing with TCA
4. Nail Fold Excision (Winograd/Frost procedures)
For cases where the nail fold is the primary offender (hypertrophied soft tissue):
- Remove the overhanging lateral nail fold
- When healed, the nail edge resembles a thumbnail with an excellent functional result
- The nail itself is not altered
5. Total Nail Ablation (Zadik procedure)
- Complete permanent removal of the nail matrix
- Indicated for severe bilateral or chronic recurrent disease
- Gives a permanently nail-free toe
Post-operative Care and Patient Education
- Non-adherent dressing + compression bandage; elevate 24 hours
- Warm soaks from day 2
- No constricting footwear for 1 week
- Nail takes 4-6 months to fully regrow
- Prevention education:
- Trim nails straight across, not curved - goal is a squared nail with corners just protruding past the hyponychium
- Wear properly fitting, wide toe-box footwear
- Avoid nail-biting/picking
- In diabetics/neuropaths: regular podiatry review
Summary of Treatment by Stage
| Stage | First-line | Definitive if fails |
|---|---|---|
| Stage I | Cotton elevation, warm soaks, gutter splint, orthonyxia | Stage II/III approach |
| Stage II | Spicule removal ± antibiotics | Partial nail avulsion ± phenolisation |
| Stage III | Surgical | Partial/total nail avulsion + matricectomy |
Sources: Campbell's Operative Orthopaedics 15e (2026); Roberts & Hedges' Clinical Procedures in Emergency Medicine; Andrews' Diseases of the Skin; Dermatology 2-Vol Set 5e; Textbook of Family Medicine 9e
Presentation and management of Neuroblastoma.
Reading File
Finding Sources
Searching PubMed
neuroblastoma management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the content needed. Let me compile a comprehensive answer.
Neuroblastoma: Presentation and Management
Definition and Overview
Neuroblastoma (NB) is the most common extracranial solid tumour of childhood, arising from primordial neural crest cells that form the adrenal medulla and sympathetic ganglia. It accounts for 8-10% of all childhood cancers and approximately 15% of all childhood cancer deaths. The tumour has a unique biology - it can spontaneously regress, mature into a benign ganglioneuroma, or behave with extreme malignancy. - Campbell-Walsh-Wein Urology; Grainger & Allison's Diagnostic Radiology
Tumour Spectrum
| Tumour | Differentiation | Schwannian Stroma | Neuroblastic Cells |
|---|---|---|---|
| Neuroblastoma | Undifferentiated | Absent/sparse | Present (immature) |
| Ganglioneuroblastoma | Intermediate | Present | Present (mixed) |
| Ganglioneuroma | Fully differentiated | Dominant | Absent |
Epidemiology
- Annual incidence: 10 per 1 million live births (USA)
- Median age at diagnosis: 19 months (familial cases: 9 months)
- 36% present in infancy; 89% are <5 years; 98% diagnosed by age 10
- >50% have metastatic disease at diagnosis
- Rare in people who do not wear shoes (analogous to the environmental component in toenail disease - but here, NB is more biologically determined)
Aetiology and Genetics
- Sporadic in >99% of cases
- Familial cases (<1%): autosomal dominant inheritance; bilateral/multifocal tumours common; predisposition genes: PHOX2B and ALK (chromosome 16p12-13)
- Sibling/offspring risk: <6%
Key Molecular Markers (with prognostic significance):
| Marker | Significance |
|---|---|
| MYCN amplification (>10 copies at 2p24) | Present in 20-25% of primary tumours; 40% of advanced-stage; adverse prognosis independent of age/stage |
| DNA ploidy/index | Hyperploidy (near-triploid, index >1) = favourable; diploid = poor response to therapy |
| Chromosome 1p deletion | Adverse outcome |
| Chromosome 11q deletion | Adverse outcome |
| Chromosome 17q gain | Adverse outcome |
| ALK mutation/amplification | Poor prognosis; targetable |
- Campbell-Walsh-Wein Urology; Mulholland & Greenfield's Surgery 7e
Pathology and Histology
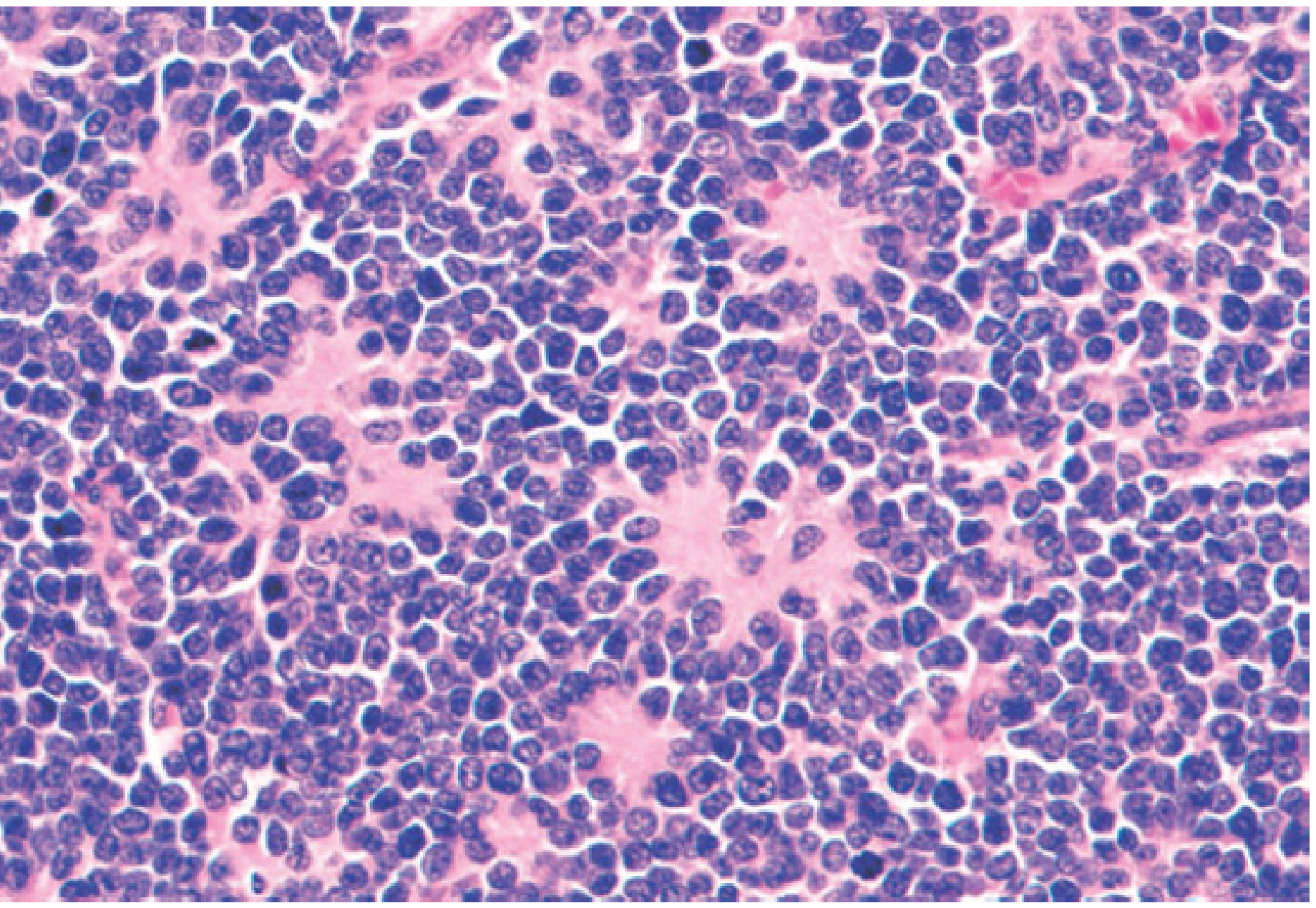
- Histology: Small, round, blue cells with hyperchromatic nuclei; Homer Wright pseudorosettes (cells palisading around blood vessels and neuritic processes) are characteristic
- Immunohistochemistry: Positive for neuron-specific enolase (NSE), synaptophysin, NB84, and tyrosine hydroxylase - distinguishes NB from other small round blue cell tumours (Ewing sarcoma, rhabdomyosarcoma, Wilms tumour)
Shimada / International Neuroblastoma Pathology Classification:
Favourable histology:
- Stroma rich, all ages, no nodular pattern
- Stroma poor, age 1.5-5 yrs, differentiated, mitosis-karyorrhexis index (MKI) <100
- Stroma poor, age <1.5 yrs (most)
Unfavourable histology:
- Stroma poor, age >5 yrs
- Stroma poor, undifferentiated (any age)
- Stroma poor, high MKI
- Nodular stroma-rich pattern
Primary Sites
| Site | Frequency |
|---|---|
| Adrenal gland | 48-50% |
| Extra-adrenal retroperitoneum (paravertebral ganglia) | 25% |
| Chest/posterior mediastinum | 16% |
| Neck | 3% |
| Pelvis (organ of Zückerkandl) | 3-4% |
75% arise in the retroperitoneum overall (adrenal + paravertebral). - Campbell-Walsh; Grainger & Allison
Clinical Presentation
Presentation is highly variable and depends on the primary site, extent of disease, and tumour biology.
By Primary Site:
Abdominal/Adrenal (most common):
- Fixed, hard abdominal mass crossing the midline
- Abdominal pain, distension
- Pelvic tumours: urinary retention, constipation (compression of bladder/bowel)
Thoracic:
- Cough, dyspnoea, respiratory symptoms
- Posterior mediastinal mass (often incidental on CXR)
- Horner syndrome (ptosis, miosis, anhidrosis) - involvement of the stellate ganglion
Cervical:
- Neck mass
- Horner syndrome
Spinal (dumbbell tumour):
- Extradural extension through neural foramina
- Spinal cord compression: weakness, paralysis, bladder/bowel dysfunction
- More common with thoracic tumours; affects up to 5% at presentation (up to 13% have radiographic evidence)
From Metastatic Disease (present in 70% at diagnosis):
- Bone pain, limping, joint pain
- Periorbital ecchymosis ("raccoon eyes") - orbital metastases; highly characteristic
- Proptosis - orbital metastases
- Anaemia, weight loss, fever
- Blueberry-muffin nodules (subcutaneous metastases) in infants
Paraneoplastic Syndromes:
| Syndrome | Mechanism | Features |
|---|---|---|
| Catecholamine excess | Tumour secretion of catecholamines | Paroxysmal hypertension, palpitations, flushing, headache, sweating (mimics phaeochromocytoma) |
| Opsoclonus-myoclonus-ataxia (OMA) | Auto-antibodies against neuroblastoma cross-reacting with normal neural tissue | Rapid multidirectional eye movements (opsoclonus), myoclonus, ataxia; associated with favourable oncologic outcome but 70-80% have prolonged neurologic sequelae |
| VIP secretion | Vasoactive intestinal peptide (VIP) | Severe watery diarrhoea (Verner-Morrison syndrome), hypokalemia |
Special Presentation - Stage 4S (MS) in infants:
- Small primary tumour, but liver, skin, and bone marrow metastases
- Tendency to spontaneous regression but risk of severe respiratory compromise from massive hepatomegaly
- Generally favourable prognosis
Diagnosis
Laboratory Evaluation
- Urinary catecholamine metabolites: Elevated in ~90% (VMA - vanillylmandelic acid; HVA - homovanillic acid; noradrenaline; dopamine); only 60% of neonates have elevated levels
- Urinary VMA and HVA: standard screening markers; if both elevated, sensitivity approaches 95%
- A positive bone marrow aspirate + elevated urinary catecholamine metabolites is sufficient to confirm diagnosis (without tissue biopsy)
- NSE (neuron-specific enolase): elevated; marker of disease burden
- LDH and ferritin: elevated in aggressive disease; adverse prognostic markers
- FBC: anaemia, thrombocytopenia (marrow infiltration)
Imaging
Initial assessment:
- Ultrasound: First-line for abdominal mass; detects calcification, heterogeneity, solid vs cystic components; also used for prenatal detection (adrenal NB on third-trimester US)
- Chest X-ray: For thoracic mass; may show calcified posterior mediastinal mass
Definitive primary tumour evaluation:
- CT or MRI: Defines tumour size, vascular encasement, lymph node involvement, intraspinal extension; MRI preferred for paraspinal tumours and spinal canal assessment
Metastatic workup:
- ¹²³I-mIBG (meta-iodobenzylguanidine) scintigraphy: Most sensitive for detecting NB metastases; uptake in 90% of tumours; used for staging, response assessment, and targeted therapy
- ⁹⁹ᵐTc-MDP bone scintigraphy: For skeletal metastases
- Bilateral bone marrow aspirate and trephine biopsy: Essential for staging
- Skeletal metastases: most common sites are skull and metaphyses of long bones (humerus, femur)
Staging
International Neuroblastoma Staging System (INSS):
| Stage | Description |
|---|---|
| 1 | Localised tumour; complete gross excision ± microscopic residual; ipsilateral lymph nodes negative |
| 2A | Localised tumour; incomplete gross excision; ipsilateral non-adherent lymph nodes negative |
| 2B | Localised tumour ± incomplete excision; ipsilateral non-adherent lymph nodes positive |
| 3 | Unresectable unilateral tumour crossing the midline ± regional lymph node involvement; OR midline tumour with bilateral lymph node involvement |
| 4 | Distant metastases to lymph nodes, bone, bone marrow, liver, skin, or other organs |
| 4S (MS) | Localised primary (stage 1, 2A, or 2B) with dissemination limited to skin, liver, and/or bone marrow; age <18 months |
International Neuroblastoma Risk Group (INRG) Staging System (pre-surgical, image-based):
- L1: Localised tumour; no image-defined risk factors (IDRFs)
- L2: Localised tumour; one or more IDRFs present
- M: Distant metastatic disease
- MS: Metastatic disease limited to skin, liver, bone marrow in children <18 months
Risk Stratification
Treatment in all cooperative group trials is based on risk group (not stage alone):
| Risk Group | Criteria | Survival |
|---|---|---|
| Low risk | Stage 1-2, favourable biology (no MYCN amp, favourable histology, hyperploid) | >95% |
| Intermediate risk | Stage 3 (favourable biology) or Stage 4S (unfavourable biology) or infant Stage 4 (no MYCN amp) | ~90-96% |
| High risk | Stage 4 (age >18 months), or any stage with MYCN amplification, or Stage 3 (unfavourable histology/biology) | ~40-50% |
Management
Management is entirely risk-stratified and multimodal.
Low-Risk Disease (Stages 1-2, favourable biology)
- Surgery alone is curative in most cases
- Complete surgical resection is the goal
- Even with microscopic or gross residual disease (stage 2, non-MYCN amplified), observation without chemotherapy is safe - spontaneous regression of residual masses is documented (32/55 patients in the German NB97 study)
- Chemotherapy reserved for symptomatic, unresectable disease
Intermediate-Risk Disease
Chemotherapy regimen (COG study):
- 4 active agents: cyclophosphamide, doxorubicin, carboplatin, etoposide
- 4 cycles (favourable biology) or 8 cycles (unfavourable biology)
- Radiotherapy only for progressive disease or unresectable primary with unfavourable features
- Surgery: gross total resection in 89%, near-total in 51 patients in one COG series - no significant difference in OS by extent of resection
- 3-year OS: 96% (98% favourable biology; 93% unfavourable biology)
High-Risk Disease (>50% of all patients)
A multimodal, sequential approach:
Phase 1: Induction Chemotherapy
- Intensive multi-agent chemotherapy (carboplatin, etoposide, vincristine, cyclophosphamide, doxorubicin - "COJEC" or COG regimens)
- Goal: maximise tumour response, downstage, convert inoperable to operable
- Surgery deferred until 13-18 weeks after chemotherapy initiation (after 3-4 courses)
- Tumours are smaller, firmer, less vascular after chemotherapy → lower risk of rupture, haemorrhage, and nephrectomy
Phase 2: Surgery (Local Control)
- Aim for >90% resection of primary tumour (associated with reduced risk of local recurrence: relative risk 0.38 for local recurrence, relative risk 0.67 for mortality vs <90% resection - LaQuaglia meta-analysis)
- Surgical complications:
- Chylous ascites (from retroperitoneal lymph node clearance)
- Chronic diarrhoea (from autonomic nerve damage around coeliac axis/SMA)
- Nephrectomy risk reduced by neoadjuvant chemotherapy
Phase 3: Consolidation (Myeloablative Therapy)
- High-dose chemotherapy + autologous stem cell transplant (ASCT)
- The ANBL0532 COG study demonstrated tandem ASCT (two sequential transplants) was superior to single ASCT
- Conditioning regimens: carboplatin + etoposide + melphalan
Phase 4: Radiotherapy
- External beam radiation to the primary tumour bed and residual disease
- Combination of gross total resection + EBRT achieves local control in 84-90%
- Radiotherapy to the spine is now avoided due to adverse effects on spinal growth
- Spinal cord compression: treat with chemotherapy first; reserve laminectomy for progressive neurological deterioration
Phase 5: Maintenance/Immunotherapy
- Isotretinoin (13-cis-retinoic acid): Promotes differentiation; given after consolidation
- Dinutuximab (anti-GD2 monoclonal antibody) + IL-2 + GM-CSF: Shown to improve event-free survival in high-risk disease (COG ANBL0032 study)
Stage 4S / Stage MS (Infants)
- Often spontaneously regresses
- Watchful waiting if asymptomatic
- Low-dose chemotherapy or low-dose radiotherapy to the liver if massive hepatomegaly threatens respiratory compromise
- Very young infants (<4 weeks) with rapid hepatomegaly are at highest mortality risk
Spinal Cord Compression (Dumbbell Tumour)
- First-line: Chemotherapy - induces rapid tumour regression
- Reserve decompressive laminectomy for progressive neurological deterioration on chemotherapy
- Radiotherapy generally avoided (adverse effects on spinal growth)
- Neurological recovery is limited in patients presenting with established severe motor deficits
Opsoclonus-Myoclonus-Ataxia Syndrome
- ACTH or corticosteroids (mainstay)
- High-dose IV immunoglobulin (IVIG)
- Cyclophosphamide ± IVIG (COG protocol)
- 70-80% have persistent neurological impairment despite oncologic cure
Prognosis - Summary
| Factor | Favourable | Unfavourable |
|---|---|---|
| Age | <18 months | >18 months |
| Stage | Low (1, 2, 4S) | High (3, 4) |
| MYCN | Not amplified | Amplified (>10 copies) |
| Histology (Shimada) | Favourable | Unfavourable |
| DNA ploidy | Hyperploid (index >1) | Diploid/tetraploid |
| Chromosomes | Normal 1p, 11q | 1p del, 11q del, 17q gain |
"Children 18 months of age or younger have a better survival rate than older children. This may be attributed to more favourable biologic parameters in tumours diagnosed at this age." - Campbell-Walsh-Wein Urology, Key Points
Key Points Summary
- Neuroblastoma is the most common extracranial solid tumour of childhood (median age 19 months)
- 75% arise in the retroperitoneum; adrenal is the most common single site
-
50% have metastatic disease at diagnosis
- Periorbital ecchymosis, Horner syndrome, and opsoclonus-myoclonus are classic paraneoplastic presentations
- Urinary VMA/HVA elevated in ~90%; combined with bone marrow aspirate, sufficient for diagnosis
- ¹²³I-mIBG scintigraphy is the gold standard for staging and response assessment
- MYCN amplification (20-25% of tumours) is the most important adverse molecular marker
- Treatment is risk-stratified: surgery alone (low-risk) → chemo + surgery (intermediate) → induction/consolidation/ASCT/immunotherapy (high-risk)
- High-risk 5-year survival: approximately 40-50% despite aggressive multimodal therapy
- Infants with stage 4S disease have an excellent prognosis, often with spontaneous regression
Sources: Campbell-Walsh-Wein Urology 3-Volume Set; Grainger & Allison's Diagnostic Radiology; Robbins & Kumar Basic Pathology; Mulholland & Greenfield's Surgery 7e; Tietz Textbook of Laboratory Medicine 7e
Neuroblastoma: Presentation and Management
I covered Neuroblastoma in detail in my previous response just above. Here is a structured summary for quick reference:
Neuroblastoma: Presentation and Management
Quick Overview
- Most common extracranial solid tumour of childhood (8-10% of all childhood cancers)
- Arises from neural crest cells of the adrenal medulla and sympathetic ganglia
- Median age at diagnosis: 19 months; 89% are <5 years old
-
50% have metastatic disease at diagnosis
- Unique biology: can spontaneously regress, mature to ganglioneuroma, or behave with extreme malignancy
Primary Sites
| Site | Frequency |
|---|---|
| Adrenal gland | 48-50% |
| Extra-adrenal retroperitoneum (paravertebral ganglia) | 25% |
| Chest / posterior mediastinum | 16% |
| Neck | 3% |
| Pelvis (organ of Zückerkandl) | 3-4% |
Presentation
By Site
| Site | Features |
|---|---|
| Abdomen | Fixed hard mass crossing midline, abdominal pain |
| Pelvis | Urinary retention, constipation |
| Thorax | Cough, dyspnoea, posterior mediastinal mass |
| Cervical/thoracic | Horner syndrome (ptosis, miosis, anhidrosis) from stellate ganglion involvement |
| Spinal (dumbbell tumour) | Spinal cord compression - weakness, paraplegia, bladder/bowel dysfunction |
From Metastatic Disease (present in 70% at diagnosis)
- Periorbital ecchymosis ("raccoon eyes") - orbital metastases; classic sign
- Proptosis - orbital metastases
- Bone/joint pain, limping
- Anaemia, weight loss, fever
- Blueberry-muffin nodules (subcutaneous metastases) in infants
- Massive hepatomegaly in Stage 4S infants → respiratory compromise
Paraneoplastic Syndromes
| Syndrome | Features | Notes |
|---|---|---|
| Catecholamine excess | Paroxysmal hypertension, palpitations, flushing, headache | Mimics phaeochromocytoma |
| Opsoclonus-myoclonus-ataxia (OMA) | Rapid multidirectional eye movements, myoclonus, ataxia | Favourable oncologic prognosis but 70-80% get permanent neurological sequelae |
| VIP secretion | Severe watery diarrhoea, hypokalemia | Verner-Morrison syndrome |
Diagnosis
Biochemical Markers
- Urinary VMA + HVA (vanillylmandelic acid + homovanillic acid): elevated in ~90%
- Positive bone marrow aspirate + elevated urinary catecholamines = sufficient for diagnosis (no tissue biopsy needed)
- Elevated NSE, LDH, ferritin = adverse markers
Imaging
| Modality | Role |
|---|---|
| Ultrasound | First-line for abdominal mass; prenatal detection |
| CT / MRI | Define primary tumour, vascular encasement, intraspinal extension |
| ¹²³I-mIBG scintigraphy | Gold standard for metastatic staging and response assessment; uptake in 90% |
| ⁹⁹ᵐTc-MDP bone scan | Skeletal metastases |
| Bilateral bone marrow aspirate + trephine | Essential for staging |
Pathology
- Small round blue cells with hyperchromatic nuclei
- Homer Wright pseudorosettes (palisading around blood vessels/neuritic processes) - characteristic
- IHC: NSE+, synaptophysin+, NB84+, tyrosine hydroxylase+ (distinguishes from Ewing sarcoma, rhabdomyosarcoma, Wilms)
Staging (INSS)
| Stage | Description |
|---|---|
| 1 | Localised; complete excision ± microscopic residual; ipsilateral nodes negative |
| 2A | Localised; incomplete excision; ipsilateral nodes negative |
| 2B | Localised; ipsilateral non-adherent nodes positive |
| 3 | Unresectable; crosses midline; OR bilateral nodal involvement |
| 4 | Distant metastases (bone, bone marrow, liver, distant nodes) |
| 4S/MS | Stage 1/2 primary + skin, liver, bone marrow mets only; age <18 months |
Prognostic Factors
| Factor | Favourable | Unfavourable |
|---|---|---|
| Age | <18 months | >18 months |
| Stage | 1, 2, 4S | 3, 4 |
| MYCN amplification | Not amplified | Amplified (>10 copies; in 20-25% of primary tumours) |
| Histology (Shimada) | Favourable | Unfavourable |
| DNA ploidy | Hyperploid (index >1) | Diploid |
| Chromosomal | Normal 1p, 11q | 1p del, 11q del, 17q gain |
Management - Risk-Stratified
Low-Risk (Stages 1-2, favourable biology) → Surgery Alone
- Complete surgical resection is curative
- Residual non-MYCN-amplified disease after surgery: observe (spontaneous regression documented)
- OS >95%
Intermediate-Risk → Chemotherapy + Surgery
- Cyclophosphamide + Doxorubicin + Carboplatin + Etoposide
- 4 cycles (favourable) or 8 cycles (unfavourable biology)
- Radiotherapy only for progressive/unresectable disease
- 3-year OS: 96%
High-Risk (>50% of all patients) → Multimodal Intensive Therapy
Sequential 5-phase approach:
| Phase | Treatment |
|---|---|
| 1. Induction | Intensive multi-agent chemotherapy (COJEC / COG regimen); 3-4 courses over 13-18 weeks |
| 2. Surgery | Resection of primary tumour; aim for >90% resection (reduces local recurrence RR 0.38; mortality RR 0.67); surgery after chemotherapy = safer, smaller, firmer tumour |
| 3. Consolidation | High-dose chemotherapy + autologous stem cell transplant (ASCT); tandem ASCT superior to single ASCT |
| 4. Radiotherapy | External beam to primary bed; laminectomy for spinal compression only if progressive neuro deficit on chemo |
| 5. Maintenance | Isotretinoin (differentiation therapy) + Dinutuximab (anti-GD2 antibody) + IL-2 + GM-CSF (immunotherapy) |
- High-risk 5-year OS: ~40-50%
Stage 4S / MS (Infants)
- Watchful waiting if asymptomatic (spontaneous regression expected)
- Low-dose chemotherapy or low-dose hepatic irradiation if massive hepatomegaly threatens breathing
Spinal Cord Compression
- Chemotherapy first (rapid tumour regression)
- Laminectomy only for progressive neurological deterioration
- Radiotherapy avoided (spinal growth impairment)
Opsoclonus-Myoclonus-Ataxia
- ACTH or corticosteroids + IVIG ± cyclophosphamide
Surgical Complications to Know
- Chylous ascites - from retroperitoneal lymph node clearance
- Chronic diarrhoea - from autonomic nerve damage (coeliac axis/SMA)
- Nephrectomy risk - reduced significantly by neoadjuvant chemotherapy
- Turned-up pulp deformity - not relevant here (this is toenail surgery!)
Sources: Campbell-Walsh-Wein Urology; Grainger & Allison's Diagnostic Radiology; Robbins & Kumar Basic Pathology; Mulholland & Greenfield's Surgery 7e
. Discuss the indications, operative steps and postoperative care of decompressive craniotomy for head trauma. [3+4+3]
Finding Sources
Searching PubMed
decompressive craniectomy traumatic brain injury
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
I now have everything needed. Here is a comprehensive, exam-formatted answer structured to the [3+4+3] mark allocation.
Decompressive Craniotomy/Craniectomy for Head Trauma
Note on Terminology: In current practice, the procedure is most precisely called decompressive craniectomy (DC) - the bone flap is permanently removed rather than replaced at the end of the operation. "Decompressive craniotomy" refers to the surgical approach where a bone flap is raised; when it is left off to allow brain expansion, it becomes a craniectomy. The terms are often used interchangeably in the context of head trauma.
Part 1: Indications [3 Marks]
Decompressive craniectomy for head trauma is indicated in two broad scenarios: mass lesion and refractory intracranial hypertension (without or after mass lesion evacuation).
A. Mass Lesion Indications (Evacuating a Clot + Leaving Bone Off)
-
Epidural haematoma (EDH)
- Volume >30 mL, OR
- Clot thickness >15 mm, OR
- Midline shift >5 mm, OR
- Associated neurological deficit
- Effacement of basal cisterns on CT
-
Acute subdural haematoma (ASDH)
- Thickness ≥1 cm on CT, OR
- Any midline shift in association with neurological decline
- Regardless of GCS, large lesions should be evacuated before deterioration
-
Intracerebral haemorrhage / large contusion
- Progressive neurological deterioration
- Refractory intracranial hypertension (ICP >20-25 mmHg)
- Midline shift with impending herniation
-
Depressed skull fracture
- Open, compound, or markedly depressed (>1 table thickness)
- Formal craniotomy for elevation, débridement, and dural repair
-
Penetrating head injury
- Debridement, haemostasis, and dural closure
B. Secondary (Decompressive) Craniectomy for Refractory Intracranial Hypertension
Performed as Tier 3 management when all medical measures have failed:
Medical management must have been tried and failed first (stepwise ICP protocol):
- Head elevation 30°
- Analgesia, sedation (propofol/morphine/midazolam)
- CSF drainage via ventriculostomy (EVD)
- Osmotherapy (mannitol 0.25-1 g/kg; hypertonic saline)
- Hyperventilation (ETCO₂ 30-35 mmHg) - short-term bridging only
- Vasopressors to maintain CPP ≥60-70 mmHg
- Barbiturate coma (if above fails)
Indication for secondary DC:
- ICP persistently >20-25 mmHg for >1 hour (or per protocol thresholds)
- Despite full first and second-tier medical management
- Within 72 hours of injury (RESCUEicp criteria: ICP >25 mmHg for 1-12 hours)
- Severe TBI with GCS ≤8, abnormal CT (diffuse swelling, midline shift)
- Unilateral fixed and dilated pupil (imminent transtentorial herniation)
Contraindications / poor prognosis factors:
-
Bilateral fixed dilated pupils (indicates irreversible brainstem herniation)
-
Unsurvivable injuries (e.g., devastating penetrating brain injury)
-
Clear evidence of brain death
-
Incompatibility with patient's goals of care
-
Sabiston Textbook of Surgery; Mulholland & Greenfield's Surgery 7e; Harrison's Principles 22e
Part 2: Operative Steps [4 Marks]
Pre-operative Preparation
- Anaesthesia: General anaesthesia; rapid-sequence intubation (RSI) with inline cervical stabilisation (assume C-spine injury until proven otherwise)
- Avoid succinylcholine-induced transient ICP rise where possible; rocuronium preferred
- Propofol (1.5-3 mg/kg) to blunt intubation-associated ICP spike
- IV access (large bore), arterial line, urinary catheter
- Reverse coagulopathy: prothrombin complex concentrate for anticoagulants; DDAVP for antiplatelet agents
- Target SpO₂ >95%, normocapnia (PaCO₂ 35-40 mmHg) intraoperatively; mild hyperventilation (PaCO₂ 30-35) only if acute herniation
Positioning
- Supine with head on headrest or horseshoe ring
- For unilateral hemicraniectomy (most common for unilateral mass lesions): head rotated contralaterally 30-45° and slightly elevated
- Mayfield skull clamp fixation in elective/semi-elective cases
- Head elevated 15-30° to facilitate venous drainage
Skin Incision (Question mark / Trauma flap)
- Large trauma flap (question mark incision): The standard approach for decompressive hemicraniectomy
- Begins just anterior to the tragus (pre-auricular), curves posteriorly behind the ear, superiorly above the pinna, and anteriorly toward the midline in a large arc
- Provides wide exposure of frontal, temporal, and parietal regions
- For bifrontal decompression (DECRA-type): Bicoronal incision from one ear to the other, behind the hairline
Craniotomy / Craniectomy
- Burr holes: Multiple burr holes placed at the corners of the proposed bone flap (typically 4-6 burr holes for a large flap) using a Hudson brace or powered drill
- Bone flap: A craniotome (power saw) is used to connect the burr holes and cut the bone flap; the flap is elevated with a periosteal elevator
- For decompressive craniectomy: The bone flap should be ≥12 cm × 12 cm (large enough to allow adequate decompression); a small craniectomy worsens outcome by causing focal pressure on the herniated brain edge
- Epidural haematoma: Clot is a liquified or solid collection between bone and dura; remove with suction and irrigation; identify and coagulate the middle meningeal artery (the source in >85%)
- Dura is opened: Cruciate or stellate dural incision to allow brain decompression
Haematoma Evacuation and Haemostasis
- Subdural haematoma: After dural opening, acute clot is evacuated with suction, irrigation (warm saline), and bipolar coagulation; underlying cerebral lacerations are débrided
- Intracerebral haemorrhage/contusion: Through a cortical incision or via a transcortical approach; aspirate haematoma, achieve haemostasis with bipolar and haemostatic agents (Surgicel, Gelfoam)
- Identify and control all bleeding points with bipolar diathermy
- Cortical bridging veins torn by trauma are coagulated and divided
Dural Augmentation (Duraplasty)
- For decompressive craniectomy, the dura must be expanded rather than simply closed under tension
- A dural patch (pericranium, fascia lata, or synthetic dural substitute) is sewn in to enlarge the dural compartment - this is the duraplasty
- The dura is sutured to the patch in a watertight fashion to prevent CSF leakage and infection
- This step is critical: closing the dura under tension negates the decompression
ICP Monitor Placement
- An external ventricular drain (EVD) or intraparenchymal Camino bolt is placed at the time of surgery
- Indicated if pre-operative GCS <8, or significant brain swelling at surgery
- EVD has the advantage of allowing therapeutic CSF drainage post-operatively
Wound Closure (Without Bone Replacement)
- Bone flap is not replaced - this is the defining feature of a craniectomy
- Bone flap is stored:
- Cryopreserved (frozen at -70°C in sterile container), OR
- Implanted subcutaneously in the patient's abdominal wall
- Temporalis muscle and galea are closed over the dural patch
- Scalp is closed in layers with staples or sutures
- The patient is left with a large bony defect covered only by scalp (the "sunken flap" appearance)
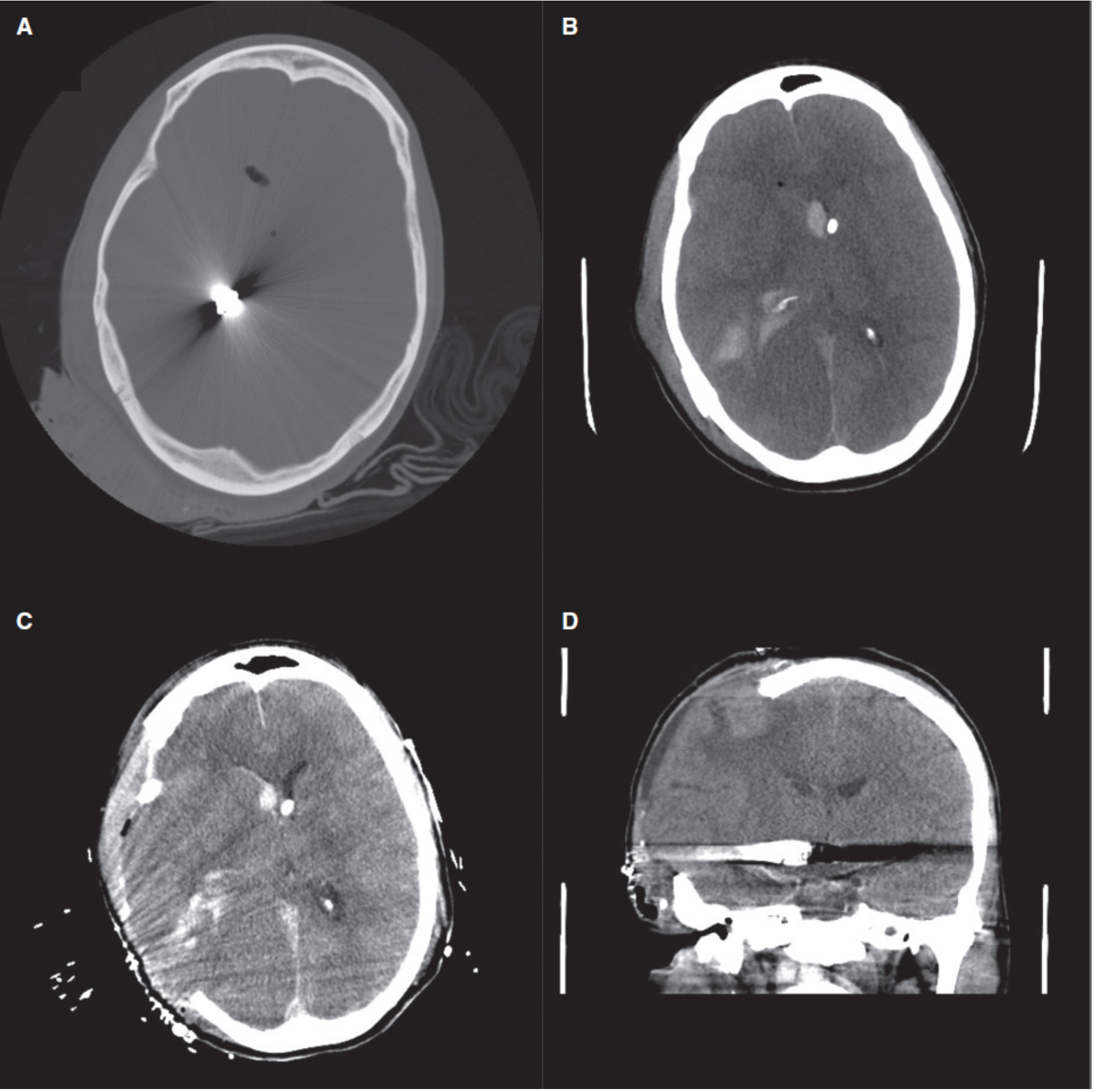
- Sabiston Textbook of Surgery; Mulholland & Greenfield's Surgery 7e
Part 3: Postoperative Care [3 Marks]
ICU Monitoring (All patients with GCS ≤8 or post-operative severe TBI)
- Neuromonitoring: Continuous ICP monitoring via EVD or intraparenchymal monitor; target ICP <20-25 mmHg and CPP 60-70 mmHg (CPP = MAP - ICP)
- Neurological observations: Hourly GCS, pupils, limb movements; prompt repeat CT on any deterioration
- Repeat CT scanning: Within 24 hours post-operatively, and on any neurological change, to identify re-accumulation of haematoma, cerebral oedema, or new haemorrhage
Respiratory and Haemodynamic Management
- Mechanical ventilation: maintain normocapnia (PaCO₂ 35-40 mmHg); avoid hypoxia (SpO₂ >95%)
- Avoid hypotension: systolic BP >90 mmHg; MAP sufficient to maintain CPP ≥60-70 mmHg
- Vasopressors (noradrenaline, phenylephrine, dopamine) as needed to support MAP
- Head of bed elevated 30° to facilitate venous drainage and reduce ICP
ICP Management (Tiered Protocol)
Tier 1:
- Head elevation 30°, analgesia, avoid hyperthermia
- EVD CSF drainage (if in situ)
Tier 2:
- Osmotherapy: Mannitol 0.25-0.5 g/kg IV 4-6 hourly (target serum osmolality 295-310 mOsm/L); OR hypertonic saline (3% NaCl)
- Sedation: propofol or midazolam; add neuromuscular blockade if necessary
- Short-term hyperventilation (PaCO₂ 30-35 mmHg) as bridge to definitive treatment
Tier 3 (if not already decompressed):
- Barbiturate coma (pentobarbital/thiopentone)
- Hypothermia (33-35°C) - note: not shown to improve survival alone
- Decompressive craniectomy (if not already performed)
Prevention and Management of Secondary Insults
- Normoglycaemia: Avoid hyperglycaemia (>180 mg/dL) and hypoglycaemia; both worsen outcome
- Normothermia: Fever increases CMRO₂ and worsens outcome; treat aggressively with paracetamol, cooling blankets
- Hyponatraemia prevention: Monitor serum Na⁺ and osmolality daily; avoid hypotonic fluids; watch for SIADH and diabetes insipidus (common with severe TBI)
- Coagulopathy: Correct ongoing coagulopathy; monitor PT/INR, platelets, fibrinogen
- Seizure prophylaxis: Phenytoin or levetiracetam for 7 days post-injury (prevents early post-traumatic seizures)
- DVT prophylaxis: Pneumatic compression stockings from day 1; pharmacological prophylaxis once intracranial haemorrhage is stable (typically 48-72 hours post-op)
Nutrition and General ICU Care
- Early enteral feeding (within 24-48 hours) via nasogastric tube; promotes gut integrity, reduces infection
- Stress ulcer prophylaxis (proton pump inhibitor or H₂ blocker)
- Strict glucose control
- Prevention of hospital-acquired infections (VAP bundle, CLABSI prevention)
- Paroxysmal sympathetic hyperactivity ("sympathetic storming") - treat with propranolol, bromocriptine, morphine
Cranioplasty (Delayed Bone Flap Replacement)
- Bone flap replacement (cranioplasty) is performed once:
- Brain swelling has resolved (typically 6-12 weeks)
- The patient is medically stable and infection-free
- The scalp wound is fully healed
- Options: original autologous bone (cryopreserved or from abdominal subcutaneous pocket), or custom titanium/PEEK implant
- Cranioplasty protects the brain, corrects cosmetic deformity, and may improve neurological function (the "syndrome of the trephined")
Evidence Base
- DECRA trial (2011): Bifrontal craniectomy vs. medical management; DC reduced mortality but increased vegetative state/severe disability at 6 months - criticised for low ICP threshold and atypical surgical approach
- RESCUEicp trial (2016): DC vs. medical management; DC improved survival; at 12 months, surgical patients had higher rates of independent living - more clinically relevant endpoint
- Both trials confirm: DC reliably lowers ICP and improves survival, but at the cost of more survivors with severe disability; careful patient selection is essential
Summary Table
| Component | Key Points |
|---|---|
| Indications | EDH >30 mL; ASDH ≥1 cm/shift; refractory ICP >20-25 mmHg (Tier 3); imminent herniation; depressed/compound fracture |
| Operative steps | RSI → position → trauma flap incision → burr holes → craniectomy ≥12 cm → dural opening → haematoma evacuation → haemostasis → duraplasty → EVD → scalp closure (bone NOT replaced) |
| Postoperative care | ICU: ICP monitoring (target <20-25 mmHg, CPP 60-70); ventilation; osmotherapy; normoglycaemia; normothermia; seizure prophylaxis; delayed cranioplasty at 6-12 weeks |
Sources: Sabiston Textbook of Surgery 21e; Mulholland & Greenfield's Surgery 7e; Harrison's Principles of Internal Medicine 22e; Morgan & Mikhail's Clinical Anaesthesiology 7e
b) Discuss the etiopathogenesis and management of case of trigeminal neuralgia. [2+3]
Finding Sources
Searching PubMed
trigeminal neuralgia treatment management
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content. Let me compile the structured exam answer.
Trigeminal Neuralgia: Etiopathogenesis and Management
Definition
Trigeminal neuralgia (TN), also called tic douloureux, is a chronic pain condition characterised by recurrent paroxysms of unilateral, sharp, lancinating, electric-shock-like pain in the distribution of one or more divisions of the trigeminal nerve (CN V). It is the most common facial pain syndrome, with an incidence of 4 per 100,000, most common between 50-70 years, and slightly more common in women (1.5:1). - Goldman-Cecil Medicine
Part 1: Etiopathogenesis [2 Marks]
Classification (ICHD-3)
| Type | Description |
|---|---|
| Classical TN | Neurovascular compression of the nerve root with morphological change (atrophy or displacement) on MRI/surgery |
| Secondary TN | Due to an identifiable underlying disease (MS, tumour) |
| Idiopathic TN | No underlying cause; no morphological nerve change |
Primary Mechanism - Neurovascular Compression
The leading hypothesis for classical TN is pulsatile compression of the trigeminal nerve root entry zone (REZ) by an adjacent vessel:
- Most common vessel: Superior cerebellar artery (SCA)
- Others: Anterior inferior cerebellar artery (AICA), posterior inferior cerebellar artery (PICA), superior petrosal vein, basilar artery (ectatic)
- Vascular contact/compression increases with age due to arterial elongation and tortuosity - explaining why TN is predominantly a disease of older adults
Pathophysiological Cascade
Vascular compression at REZ
↓
Focal demyelination of primary trigeminal afferents
↓
Loss of insulation between adjacent sensory fibres
↓
Ephaptic (cross-talk) transmission between fibres
↓
Focal hyperexcitability + ectopic neuronal discharges
↓
Paroxysmal pain triggered by innocuous stimuli
- Pathology studies confirm: vacuolated neurons, segmental demyelination, and vascular changes in the gasserian ganglion of TN patients vs. controls
- The REZ (where central and peripheral myelin meet - the Obersteiner-Redlich zone) is particularly vulnerable because peripheral myelin (Schwann cell-derived) is less robust than central myelin (oligodendrocyte-derived)
Secondary Causes
- Multiple sclerosis (demyelinating plaque at REZ) - TN in younger patients (<40 yrs) should prompt MRI for MS
- Posterior fossa tumours: Meningioma, acoustic neuroma (schwannoma), epidermoid cyst
- Vascular malformations: AVM, aneurysm
- Skull base malignancy: Nasopharyngeal carcinoma invading skull base
- Pontine lacunar infarct affecting the trigeminal nucleus
Sensory loss or motor weakness of the trigeminal nerve on examination suggests a secondary cause (space-occupying lesion or neuropathy), not classical TN. Classical TN has a normal neurological examination. - Bradley & Daroff's Neurology
Rare/Familial Cases
A small number of familial cases suggest a genetic predisposition; the exact gene has not been conclusively identified.
Part 2: Management [3 Marks]
Management follows a stepwise approach: medical → interventional → surgical.
A. Medical (Pharmacological) Management
First-Line Drugs (Sodium Channel Blockers)
| Drug | Dose | Notes |
|---|---|---|
| Carbamazepine | 100-400 mg BD/TDS (up to 1200 mg/day) | Drug of choice; effective in 70-80%; monitor LFTs, FBC; SIADH risk |
| Oxcarbazepine | 300-600 mg BD | Better tolerated than carbamazepine; fewer drug interactions |
Second-Line Drugs
| Drug | Notes |
|---|---|
| Gabapentin | Benign side-effect profile; useful alternative or adjunct |
| Pregabalin | Similar to gabapentin |
| Phenytoin | IV fosphenytoin 15-20 mg PE/kg for acute severe attacks |
| Baclofen | GABA-B agonist; useful as adjunct |
| Lamotrigine | Especially useful in MS-associated TN |
| Valproate, clonazepam, topiramate | Third-line alternatives |
Acute severe attacks: IV fosphenytoin or local anaesthesia of the conjunctival sac with proparacaine ophthalmic drops (provides relief for hours to days). - Bradley & Daroff's
B. Interventional (Percutaneous) Procedures
For patients with medically refractory disease or drug intolerance:
1. Peripheral Nerve Block / Alcohol Injection
- Infraorbital nerve (V2), supraorbital nerve (V1), mental/mandibular nerve (V3)
- 0.5-0.75 mL absolute alcohol injected
- Pain relief in high proportion, but relapse within 6-18 months
- Advantages: low morbidity, temporary sensory loss
- Can be repeated 1-2 times; thereafter more proximal procedures needed
2. Percutaneous Radiofrequency Thermocoagulation (RFT)
- Needle passed through foramen ovale into the gasserian ganglion
- Radiofrequency current produces selective thermal injury to pain fibres
- Pain relief in up to 93%; procedure can be repeated on relapse
- Procedure of choice for elderly or medically unfit patients
- Complications: carotid artery injury, motor root damage (jaw weakness), corneal anaesthesia (dangerous in V1 disease - risk of neuroparalytic keratitis), anaesthesia dolorosa (post-traumatic trigeminal neuropathy with painful numbness)
3. Percutaneous Glycerol Rhizotomy
- Injection of glycerol into Meckel's cave (the trigeminal cistern) via foramen ovale
- Chemically injures the nerve root
- Effective but higher recurrence rate than RFT
4. Percutaneous Balloon Compression
- Fogarty balloon inflated within Meckel's cave, compressing the ganglion
- Technically simple; effective
- Higher early recurrence rate (pain recurs 2-3 years later)
5. Stereotactic Radiosurgery (Gamma Knife / LINAC)
- Focused radiation (70-90 Gy) delivered precisely to the trigeminal REZ
- Non-invasive; ideal for poor surgical candidates
- Onset of relief: delayed (weeks to months)
- Relatively high recurrence rate; risk of facial numbness (dysaesthesia) in up to 30-50%
- Less favoured than MVD as first definitive surgical option
C. Surgical Management - Microvascular Decompression (MVD) (Jannetta Procedure)
The only procedure that addresses the underlying cause and is potentially curative.
Indications:
- Medically refractory TN
- Classical TN with documented neurovascular compression on MRI
- Young, fit patients (good surgical candidates)
- Failure or relapse after percutaneous procedures
Technique:
- Retromastoid/suboccipital craniotomy (small posterior fossa craniotomy behind the ear on the symptomatic side)
- The cerebellum is gently retracted
- The trigeminal nerve root is explored at its entry into the pons
- The offending vessel (usually the SCA) is identified, freed from adhesions
- A Teflon felt pledget or other non-absorbable material is interposed between the vessel and the nerve root, padding the vessel away from the nerve
Results:
- Initial pain relief: 85-90%
- Long-term pain-free rates (5-10 years): 70-80%
- Lowest recurrence rate of all surgical options
- Preserves facial sensation (major advantage over percutaneous rhizotomy)
Complications:
- Cerebellar injury
- Hearing loss (VIII nerve proximity)
- Facial weakness (VII nerve)
- CSF leak
- Meningitis
- Mortality: <0.5% in experienced centres
"MVD remains the first definitive management option because SRS is associated with a substantial incidence of facial numbness." - Schwartz's Principles of Surgery 11e
Summary: Management Algorithm
Trigeminal Neuralgia
↓
MEDICAL THERAPY (First-line)
Carbamazepine / Oxcarbazepine
↓ (failure / intolerance)
SECOND-LINE DRUGS
Gabapentin, Baclofen, Lamotrigine, Phenytoin
↓ (refractory)
INTERVENTIONAL / SURGICAL
↙ ↘
Young, fit patient Elderly/unfit patient
with NVC on MRI or patient preference
↓ ↓
Microvascular Percutaneous RFT /
Decompression (MVD) Glycerol / Balloon
[Curative, preserves Gamma Knife SRS
sensation; gold standard]
Sources: Bradley & Daroff's Neurology in Clinical Practice; Goldman-Cecil Medicine; Schwartz's Principles of Surgery 11e; Rosen's Emergency Medicine
Briefly discuss the mechanism, clinical presentation, diagnosis and management of chronic subdural hematoma. [2+2+2+4]
Finding Sources
Searching PubMed
chronic subdural hematoma management treatment
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
I now have all the content needed for a complete, exam-structured answer.
Chronic Subdural Haematoma (CSDH)
Definition
Chronic subdural haematoma (CSDH) is defined as a collection of blood and its breakdown products in the subdural space, with symptoms developing 2 weeks or more after the initiating event. It differs fundamentally from acute SDH in its pathogenesis, CT appearance, consistency, and surgical approach. - Goldman-Cecil Medicine
Part 1: Mechanism (Etiopathogenesis) [2 Marks]
Predisposing Factors (the "at-risk brain")
CSDH most commonly affects patients whose brain is atrophied, creating a larger subdural space and stretching the bridging veins:
- Age >65 years (cerebral atrophy)
- Alcoholism (cerebral atrophy + coagulopathy)
- Anticoagulant or antiplatelet therapy (warfarin, DOACs, aspirin, clopidogrel)
- Chronic haemodialysis (heparin use + uraemic platelet dysfunction)
- Intracranial hypotension (CSF leaks, VP shunts - brain descent stretches bridging veins)
Initiating Event - Bridging Vein Tear
- Minor head trauma (often trivial and forgotten - no history in ~50% of patients)
- The atrophic, mobile brain shifts within the skull
- Cortical bridging veins (running from cortex to dural sinuses) are stretched and torn
- A small acute subdural haematoma forms
Membrane Formation
- The initial bleed is surrounded by a fibrous capsule (pseudomembrane) that grows inward from the dura
- An outer membrane (thick, adherent to dura, highly vascular - from dural fibroblasts and granulation tissue)
- An inner membrane (thin, adherent to arachnoid)
Mechanism of Enlargement (Three Complementary Hypotheses)
| Mechanism | Description |
|---|---|
| Re-bleeding from neomembranes | The outer membrane contains thin-walled, fragile sinusoidal capillaries that lack tight junctions; these leak blood and fibrinolytic products repeatedly → progressive enlargement |
| Osmotic expansion (Gardner, 1932) | Breakdown of blood into smaller molecules (haemoglobin → bilirubin → smaller proteins) raises osmotic pressure → draws fluid from surrounding tissues into the clot cavity |
| Inflammatory exudation | Breakdown products of blood elements trigger inflammatory mediators, VEGF, and angiogenic factors → increased capillary permeability → further fluid accumulation |
The current favoured hypothesis (Goldman-Cecil 2024): Initial mechanical insult splits the dural border cell layer → reactive inflammation → release of angiogenic factors → fragile neovascular membrane → repeated exudation of blood, fibrinolytic factors, and inflammatory mediators
- The larger the initial clot, the more likely it is to enlarge (critical volume threshold)
- Fibrinolytic factors within the haematoma maintain it in a liquid state and promote re-bleeding
Part 2: Clinical Presentation [2 Marks]
CSDH is the "great masquerader" - it mimics numerous neurological and psychiatric conditions. The classical triad of trauma, lateralising signs, and altered consciousness is often absent.
Typical Patient
- Elderly person with cerebral atrophy
- History of trivial or forgotten head injury weeks prior
- Gradual, progressive symptoms over days to weeks with fluctuating course
Symptoms (in order of frequency)
| Symptom | Notes |
|---|---|
| Headache (~70%) | Usually dull, persistent, worse in mornings |
| Altered mental status | Confusion, drowsiness, apathy, slowness of thinking - mimics dementia |
| Fluctuating consciousness | Characteristic waxing and waning level of alertness |
| Unsteady gait | Non-specific |
| Seizures | Particularly in alcoholics |
| New neurological deficits | Hemiparesis, aphasia |
Signs
- Hemiparesis - contralateral (ipsilateral in 10% due to Kernohan-Woltman notch sign - compression of contralateral cerebral peduncle against tentorial edge)
- Ipsilateral pupil dilatation - indicates uncal herniation (unreliable indicator of side in 10% of cases)
- Papilloedema - from raised ICP
- Symptoms mimicking TIA (fluctuating focal deficits)
- Bilateral CSDH (20% of cases): bilateral leg weakness from medial frontal lobe compression
- In infants: macrocrania, vomiting, convulsions
Common Misdiagnoses (before CSDH discovered)
Cerebral infarction, dementia, encephalitis, metabolic encephalopathy, depression, drug intoxication - reflecting the non-specific nature of the presentation. - Adams & Victor's Principles of Neurology
Part 3: Diagnosis [2 Marks]
Non-contrast CT Head (First-line, Gold Standard)
The CT appearance changes with haematoma age, which is diagnostically important:
| Age | CT Appearance | Explanation |
|---|---|---|
| Acute (<72 hours) | Hyperdense (white) | Fresh blood with intact proteins |
| Subacute (3 days - 3 weeks) | Isodense to brain | Haemoglobin breakdown; may be missed |
| Chronic (>3 weeks) | Hypodense (dark/black) | Iron metabolised, protein concentration drops |
| Mixed/Acute-on-chronic | Mixed density (hyperdense area in dependent portion within hypodense collection) | Re-bleeding into a chronic haematoma |
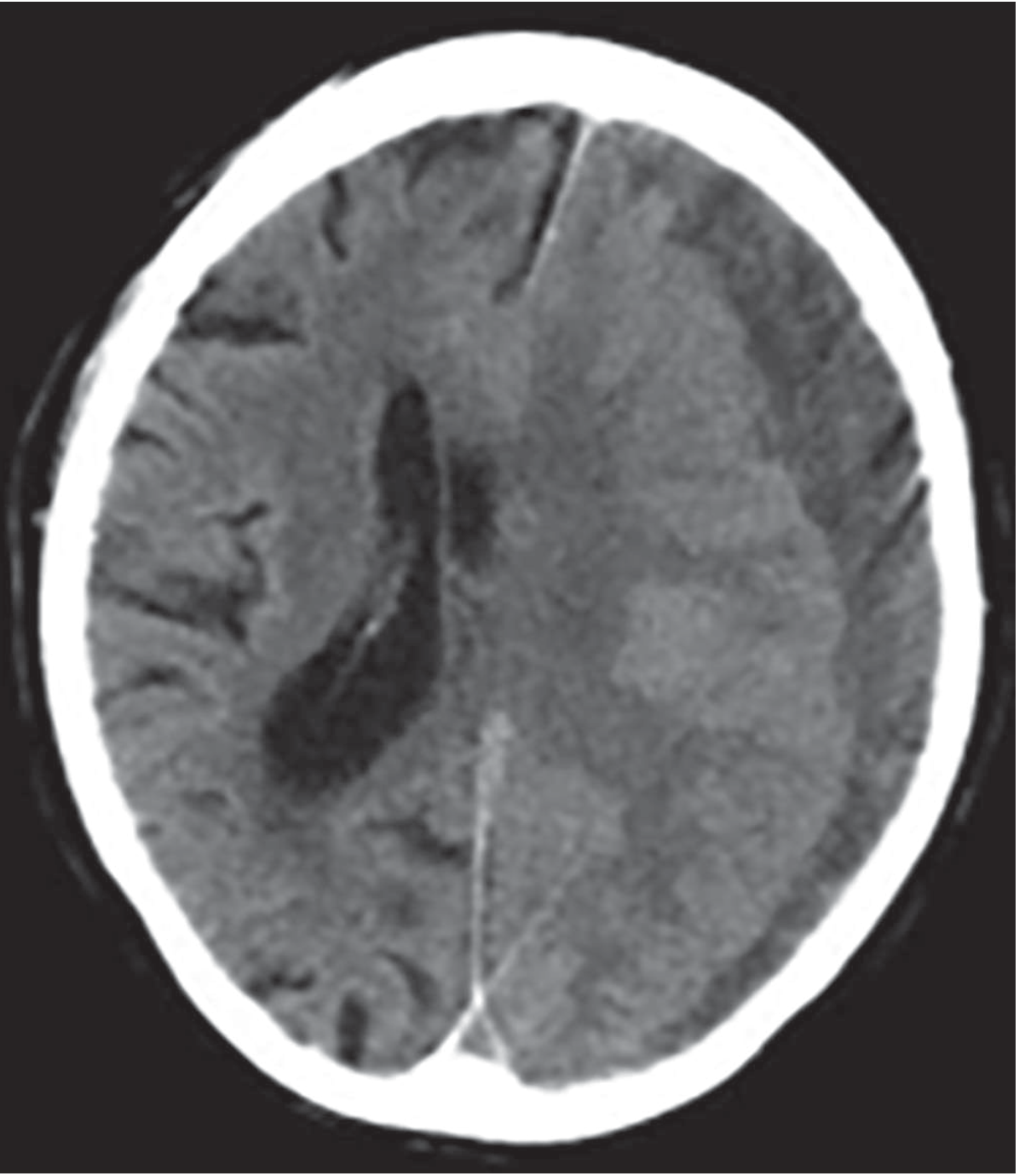
CT features to report:
- Crescent-shaped collection following the inner table of skull
- Midline shift (note direction and degree in mm)
- Effacement of sulci and ventricles
- Bilateral collections (20%)
MRI Head
- More sensitive than CT, especially for:
- Isodense (subacute) haematomas that can be missed on CT
- Bilateral isodense collections (where midline shift may be absent)
- Posterior fossa and interhemispheric collections
- CSDH: hypointense on T1, hyperintense on T2 (fluid signal)
Isodense CSDH on CT - Clues to Not Miss It
- Absence of cortical sulci on one or both sides (sulci compressed)
- Inward displacement of the grey-white matter interface
- Asymmetry of ventricles or midline shift without obvious lesion
- IV contrast CT: enhancing membranes outline the haematoma
Part 4: Management [4 Marks]
A. Conservative (Non-operative) Management
Indications:
- Asymptomatic patients
- Small collection (<1 cm) without significant mass effect
- Mild symptoms with no neurological deficit
Measures:
- Serial clinical and CT monitoring
- Stop anticoagulants/antiplatelets (with appropriate bridging if needed)
- Reverse coagulopathy (prothrombin complex concentrate for warfarin)
- Adequate hydration and nutrition
- Avoid hypotension (maintains cerebral perfusion)
Corticosteroids are not beneficial and are associated with higher morbidity. Prophylactic anti-epileptic drugs are not recommended. - Goldman-Cecil Medicine 2024
B. Surgical Management
Indications:
- CSDH >1 cm in thickness on CT
- Any symptomatic CSDH (headache, confusion, hemiparesis, deteriorating GCS)
- Midline shift with clinical features
- Progressive neurological deterioration
The consistency of CSDH is key: unlike acute SDH (thick congealed clot requiring craniotomy), CSDH is a viscous liquid - dark brown, resembling motor oil - which can be drained through a burr hole.
Procedure 1: Burr Hole Drainage (First-line; Procedure of Choice)
- Burr hole placed over the dependent (lower) edge of the collection, usually in the parietal region
- The dura is opened; the outer membrane is opened carefully
- The dark brown liquid haematoma is aspirated gently
- The cavity is copiously irrigated with warm saline (body temperature - superior to room temperature irrigation in reducing recurrences) until the fluid runs clear
- A second, more anterior burr hole may be placed if the collection is loculated by membranes and does not drain satisfactorily
- A subdural drain is left in situ for 24-48 hours post-operatively (reduces recurrence risk)
Post-operative positioning:
- Patient nursed flat (unlike other neurosurgical procedures where head-up is standard)
- This encourages brain re-expansion and prevents subdural air (pneumocephalus) from obstructing drainage
Procedure 2: Formal Craniotomy
Reserved for:
- CSDH too congealed for irrigation
- Complex membranes preventing effective drainage through burr hole
- Persistent haemorrhage not controlled through burr hole
- Recurrent CSDH after repeated burr hole drainage
- Acute-on-chronic SDH requiring clot evacuation
Procedure 3: Twist-drill / Minicraniotomy
- 5 mm twist-drill craniostomy under local anaesthesia
- Suitable for elderly, frail patients; can be performed at bedside
- Lower risk but less effective for large collections
C. Management of Recurrence (10-20% of cases)
- Serial CT at 1 month post-op
- Repeat burr hole drainage if symptomatic recollection
- Options under investigation for recurrence prevention:
- Middle meningeal artery (MMA) embolization - recent RCTs (EMPROTECT 2025, NEJM 2024) investigating its role in reducing recurrence; now emerging as adjuvant therapy
- Antifibrinolytic agents (tranexamic acid)
- Statins (dexamethasone controversy - shown to reduce recurrence but also increase complications)
D. Prognosis
- Mortality: <30%
- >80% of survivors have good or complete functional recovery at 6 months
- Poor outcome predictors: pre-op coma, age >80, bilateral collections, delayed surgery
Summary Table
| Aspect | Key Points |
|---|---|
| Mechanism | Bridging vein tear → acute SDH → membrane formation → re-bleeding from fragile neomembrane capillaries + osmotic expansion → progressive enlargement |
| Presentation | Elderly patient; trivial/forgotten trauma; headache, fluctuating confusion, hemiparesis; mimics stroke/dementia |
| Diagnosis | CT: hypodense (dark) crescent-shaped collection ± midline shift; MRI for isodense lesions |
| Management | Burr hole drainage (first-line) + subdural drain; craniotomy for complex cases; nurse flat post-op; MMA embolisation emerging for recurrence |
Sources: Adams & Victor's Principles of Neurology 12e; Goldman-Cecil Medicine 2024; Schwartz's Principles of Surgery 11e; Plum & Posner's Diagnosis and Treatment of Stupor and Coma
This is a shared conversation. Sign in to Orris to start your own chat.