Give me a very high-yield summary of hereditary spherocytosis, only the pathology aspect, for NEET PG 2026. It should cover almost 100% of high-yield topics that can be asked in interviews of NEET PG 2026.
hereditary spherocytosis pathology membrane defect spectrin ankyrin band 3 protein
hereditary spherocytosis osmotic fragility spleen sequestration hemolysis pathogenesis
hereditary spherocytosis peripheral blood smear spherocytes
hereditary spherocytosis morphology gross microscopy spleen RBC shape pathological features
hereditary spherocytosis genetics mutation inheritance autosomal dominant recessive SPTA1 SPTB ANK1 SLC4A1 EPB42
Hereditary Spherocytosis — Pathology (NEET PG 2026)
1. Basic Definition & Epidemiology
| Feature | Detail |
|---|---|
| Type | Hereditary hemolytic anemia (corpuscular, intracorpuscular defect) |
| Prevalence | ~1:2000–1:5000 in Europeans (most common hereditary hemolytic anemia in this group) |
| Credited to | Minkowski and Chauffard (end of 19th century) |
| Key feature | Presence of spherocytes in peripheral blood + increased osmotic fragility |
2. Genetics — HIGH YIELD
| Gene | Protein Encoded | Inheritance |
|---|---|---|
| ANK1 | Ankyrin-1 | AD (most common mutation, ~50–65%) |
| SPTA1 | α-Spectrin | AR (severe disease) |
| SPTB | β-Spectrin | AD |
| SLC4A1 (Band 3 gene) | Band 3 protein (Anion exchanger 1) | AD |
| EPB42 | Protein 4.2 | AR (common in Japanese) |
- Inheritance: Predominantly Autosomal Dominant (~75%), but can be AR or de novo mutation (~25%)
- Genetic heterogeneity: same clinical phenotype arises from mutations in different genes
3. Pathogenesis — THE CORE CONCEPT
RBC Membrane Structure (Know This!)
- Lipid bilayer (outer) — linked to
- Membrane skeleton (inner) — spectrin, actin, ankyrin, band 3, protein 4.1, protein 4.2
Lipid Bilayer
↕ (vertical interaction via Ankyrin–Band3–Spectrin)
Spectrin–Actin skeleton (horizontal interactions)
Defect in HS
- Vertical interaction between the lipid bilayer and the underlying cytoskeleton is disrupted
- Proteins involved: Ankyrin, Band 3, Protein 4.2 (connect bilayer to skeleton)
- Result: Loss of lipid bilayer membrane (membrane vesiculation) → reduced surface area-to-volume ratio
Why Spherocytes Form?
Normal RBC = biconcave disc (large surface area, flexible) HS RBC = spherocyte (minimum surface area for the same volume, rigid)
4. Role of the Spleen — MOST HIGH-YIELD PATHOLOGY TOPIC
Spherocyte enters splenic sinusoids
↓
Cannot deform (rigid, ↓ surface area-to-volume ratio)
↓
Trapped in splenic cords (of Billroth)
↓
Exposed to hostile microenvironment:
• Low pH
• Low glucose
• Low O₂
• Macrophage contact
↓
Further membrane loss → more spherical
↓
Extravascular hemolysis (RES/macrophage phagocytosis)
↓
Splenomegaly
- Hemolysis is extravascular (predominantly in spleen)
- Splenectomy is curative (removes site of destruction) — does NOT correct the membrane defect
- After splenectomy: spherocytes persist but hemolysis stops
5. Gross Pathology
| Organ | Finding |
|---|---|
| Spleen | Splenomegaly (congestive; red pulp expansion; congested splenic cords) |
| Liver | May be enlarged (extramedullary hematopoiesis in chronic cases) |
| Gallbladder | Pigment (bilirubin) gallstones — due to chronic hemolysis → ↑ unconjugated bilirubin |
| Bone marrow | Erythroid hyperplasia (compensatory) → "hair-on-end" skull X-ray (less pronounced than thalassemia) |
| Long bones | Expansion of medullary cavity (in severe, untreated cases) |
6. Microscopic Pathology
Peripheral Blood Smear — CLASSIC EXAM IMAGE
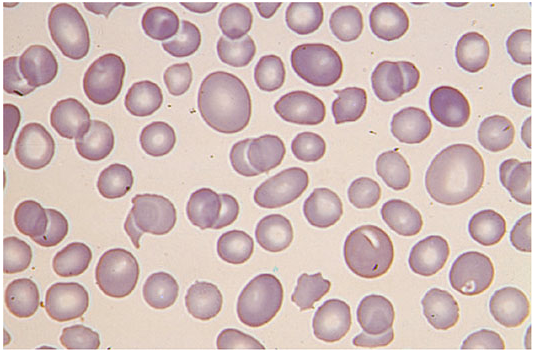
| Feature | Normal RBC | Spherocyte in HS |
|---|---|---|
| Shape | Biconcave disc | Spherical |
| Central pallor | Present (1/3 of cell) | Absent (no central pallor) |
| MCV | Normal | Normal or slightly low |
| MCHC | 32–36 g/dL | ↑ MCHC (>36 g/dL) — MOST SPECIFIC finding |
| Size | Uniform | Smaller than normal RBCs |
- No central pallor — spherocytes appear uniformly dark pink
- Microspherocytes — smaller than normal RBCs
- MCHC elevated — due to cellular dehydration (water loss follows membrane loss)
- Reticulocytosis — compensatory (polychromasia on smear)
Spleen Histology
- Congested red pulp
- Dilated splenic cords (of Billroth) packed with trapped spherocytes and macrophages
- Erythrophagocytosis by macrophages
- White pulp relatively preserved initially
Bone Marrow
- Normoblastic erythroid hyperplasia (↑ erythroid:myeloid ratio)
- In aplastic crises (Parvovirus B19): erythroid hypoplasia
7. Pathological Consequences / Complications
| Complication | Pathological Basis |
|---|---|
| Hemolytic anemia | Extravascular hemolysis in spleen |
| Jaundice (unconjugated) | ↑ Bilirubin from hemolysis |
| Pigment gallstones | ↑ Unconjugated bilirubin → precipitates as calcium bilirubinate |
| Splenomegaly | Chronic trapping and destruction of spherocytes |
| Aplastic crisis | Parvovirus B19 infects erythroid precursors → sudden ↓ in RBC production |
| Megaloblastic crisis | Folate deficiency (high demand due to erythroid hyperplasia) |
| Hemolytic crisis | Triggered by infections → sudden ↑ in hemolysis |
| Leg ulcers | Rare; due to vascular sludging |
8. Key Diagnostic Tests (Pathology-Relevant)
| Test | Finding | Mechanism |
|---|---|---|
| Osmotic Fragility Test (OFT) | Increased (spherocytes lyse at higher NaCl concentration) | ↓ Surface area-to-volume ratio; less reserve to swell before lysis |
| Incubated OFT (37°C, 24h) | More sensitive than non-incubated | Spherocytes worsen with incubation |
| Autohemolysis test | ↑ Autohemolysis; corrected by glucose (Type I pattern) | Distinguishes from G6PD |
| EMA (Eosin-5-maleimide) binding test | ↓ EMA fluorescence | EMA binds Band 3 and Rh-associated proteins; deficiency = ↓ binding |
| SDS-PAGE (gel electrophoresis) | Quantifies individual membrane proteins | Gold standard for protein identification |
| Coombs test (DAT) | Negative | No immune mechanism; helps differentiate from AIHA |
| MCHC | >36 g/dL | Most reliable routine lab finding |
EMA test is now considered the best screening test (flow cytometry-based, sensitive and specific)
9. Comparison: Key Membrane Proteins
| Protein | Location | Function | HS Subtype |
|---|---|---|---|
| Spectrin (α, β) | Skeleton | Horizontal lattice; maintains shape | Severe AR (α) or mild-moderate AD (β) |
| Ankyrin | Links spectrin to Band 3 | Vertical interaction | Most common AD form |
| Band 3 | Transmembrane | Anion exchange + anchors skeleton | AD; also reduces protein 4.2 |
| Protein 4.2 | Links ankyrin to Band 3 | Vertical stabilization | AR; Japanese population |
| Protein 4.1 | Links spectrin to actin | Horizontal interaction | NOT typically associated with HS |
10. One-Liners for Rapid Recall
- Most common hereditary hemolytic anemia in Europeans → HS
- Most common mutation → Ankyrin (ANK1)
- Most common inheritance → Autosomal Dominant
- Defect type → Vertical interaction defect → membrane vesiculation → ↓ surface area
- Hallmark lab finding → ↑ MCHC (>36 g/dL) + spherocytes without central pallor
- Diagnostic test (classic) → Osmotic fragility test (increased)
- Best modern screening test → EMA binding (flow cytometry)
- Gold standard protein identification → SDS-PAGE
- Coombs test → Negative (non-immune hemolysis)
- Hemolysis site → Extravascular (spleen, splenic cords of Billroth)
- Aplastic crisis cause → Parvovirus B19
- Megaloblastic crisis cause → Folate deficiency
- Splenectomy → Curative for hemolysis, does NOT fix membrane defect
- Gallstones type → Pigment stones (calcium bilirubinate)
- Protein 4.2 deficiency → AR, common in Japanese
- Credited to → Minkowski and Chauffard
11. Summary Flowchart
Gene mutation (ANK1 / SPTB / SLC4A1 / EPB42 / SPTA1)
↓
Defective vertical interaction (Ankyrin–Band3–Spectrin)
↓
Membrane vesiculation → Loss of lipid bilayer fragments
↓
↓ Surface area-to-volume ratio → SPHEROCYTE formation
↓
Spherocytes → Trapped in splenic cords (rigid, cannot deform)
↓
Macrophage phagocytosis → Extravascular hemolysis
↓
↑ Unconjugated bilirubin → Jaundice + Pigment gallstones
Erythroid hyperplasia → Marrow expansion
Splenomegaly → Splenectomy = cure
Okay, now convert the above summary to an Anki card with the front showing hereditary spherocytosis and the back showing all the entire details with proper spacing so that one can easily read and separate the headings. It should all be in paragraph format, the back portion. Give it to me in a text format so that I can copy it to a text file and then import.
| as a common custom separator, and HTML for formatting on the back. Copy everything between the triple-backtick blocks into a .txt file and import with Field separator: |, and allow HTML in fields.Hereditary Spherocytosis — Complete Pathology Summary (NEET PG 2026)|<b style="font-size:1.2em;">HEREDITARY SPHEROCYTOSIS — PATHOLOGY (NEET PG 2026)</b><br><br><b>BASIC DEFINITION & EPIDEMIOLOGY</b><br>Hereditary spherocytosis (HS) is a hereditary hemolytic anemia caused by an intracorpuscular (corpuscular) defect in the RBC membrane. Its prevalence is approximately 1:2000–1:5000 in populations of European ancestry, making it the most common hereditary hemolytic anemia in this group. It was first described by <b>Minkowski and Chauffard</b> at the end of the 19th century, who reported families with spherocytes in their peripheral blood. The hallmark in vitro finding was increased susceptibility to lysis in hypotonic media, which became the basis of the osmotic fragility test.<br><br><b>GENETICS</b><br>HS is genetically heterogeneous. The most common mutation is in the <b>ANK1</b> gene encoding <b>Ankyrin-1</b>, accounting for ~50–65% of cases and inherited in an <b>autosomal dominant (AD)</b> pattern. <b>SPTB</b> (β-Spectrin) is also AD. <b>SPTA1</b> (α-Spectrin) is <b>autosomal recessive (AR)</b> and causes severe disease. <b>SLC4A1</b> (Band 3 protein / Anion exchanger 1) is AD. <b>EPB42</b> (Protein 4.2) is AR and is particularly common in the <b>Japanese population</b>. Overall, ~75% of cases are AD and ~25% are AR or de novo mutations.<br><br><b>PATHOGENESIS</b><br>The RBC membrane consists of an outer lipid bilayer connected to an inner cytoskeletal scaffold (spectrin, actin, ankyrin, band 3, protein 4.1, protein 4.2). The key connections are <b>vertical interactions</b> — Ankyrin bridges Band 3 (transmembrane) to Spectrin (skeleton). In HS, these vertical interactions are disrupted. The lipid bilayer loses its attachment to the skeleton and undergoes <b>membrane vesiculation</b> — small fragments of lipid bilayer are shed. This progressively reduces the <b>surface area-to-volume ratio</b> of the RBC. A normal RBC is a biconcave disc with a large surface area and flexibility. As surface area is lost, the cell can no longer maintain its biconcave shape and becomes a <b>sphere (spherocyte)</b> — the geometric shape with the minimum surface area for a given volume. Spherocytes are rigid and cannot deform.<br><br><b>ROLE OF THE SPLEEN</b><br>Spherocytes enter the splenic microcirculation but cannot deform enough to squeeze through the narrow fenestrations of the splenic sinusoids. They become <b>trapped in the splenic cords of Billroth</b>. Within the cords, the hostile microenvironment (low pH, low glucose, low O₂, macrophage proximity) causes further membrane damage and additional sphering. Eventually, macrophages phagocytose these cells, resulting in <b>extravascular hemolysis</b>. This cycle of trapping and destruction drives progressive <b>splenomegaly</b>. Splenectomy removes the primary site of destruction and is <b>curative for the hemolysis</b>, even though the underlying membrane defect persists — spherocytes remain in circulation post-splenectomy but are no longer destroyed.<br><br><b>GROSS PATHOLOGY</b><br>The spleen is enlarged (splenomegaly) with congested red pulp and dilated splenic cords. The liver may also be enlarged due to extramedullary hematopoiesis in chronic or severe cases. The gallbladder commonly contains <b>pigment (calcium bilirubinate) gallstones</b>, resulting from chronically elevated unconjugated bilirubin. The bone marrow shows expansion of the erythroid compartment (compensatory erythroid hyperplasia), and in severe long-standing cases, expansion of the medullary cavity in long bones may occur.<br><br><b>MICROSCOPIC PATHOLOGY</b><br>On the peripheral blood smear, the hallmark is the presence of <b>spherocytes</b> — small, densely staining red cells with <b>absent central pallor</b> (normally central pallor occupies one-third of the cell). They appear uniformly hyperchromic (dark pink). <b>Reticulocytosis</b> (polychromasia) is present as a compensatory response. The <b>MCHC is elevated above 36 g/dL</b>, which is the most reliable and specific routine laboratory finding in HS; it results from cellular dehydration as water is lost along with the shed membrane. The MCV is normal or slightly reduced. In the spleen, histology reveals congested red pulp with dilated cords of Billroth packed with trapped spherocytes and macrophages actively engaged in erythrophagocytosis. The bone marrow shows normoblastic erythroid hyperplasia with an increased erythroid-to-myeloid ratio. In aplastic crisis (triggered by Parvovirus B19), there is erythroid hypoplasia with characteristic giant pronormoblasts.<br><br><b>MEMBRANE PROTEINS — KEY COMPARISON</b><br><b>Spectrin (α and β)</b> forms the horizontal lattice of the cytoskeleton and maintains the cell shape. α-Spectrin deficiency (AR) causes severe HS; β-Spectrin (AD) causes mild-to-moderate disease. <b>Ankyrin</b> is the most commonly deficient protein, linking Spectrin to Band 3, and its deficiency is the most frequent cause of AD HS. <b>Band 3</b> (SLC4A1) is a transmembrane protein serving dual roles — anion exchange and cytoskeletal anchoring; its loss also secondarily reduces Protein 4.2. <b>Protein 4.2</b> links Ankyrin to Band 3 and stabilizes vertical interactions; its deficiency follows AR inheritance and is the predominant form in Japan. <b>Protein 4.1</b> mediates horizontal spectrin-actin interactions and is NOT typically implicated in HS.<br><br><b>DIAGNOSTIC TESTS</b><br>The <b>osmotic fragility test (OFT)</b> is the classic diagnostic test — spherocytes lyse at higher NaCl concentrations than normal RBCs because their reduced surface area-to-volume ratio leaves no reserve to swell before rupturing. The <b>incubated OFT</b> (24h at 37°C) is more sensitive. The <b>autohemolysis test</b> shows increased autohemolysis that is <b>corrected by glucose</b> (Type I pattern), which helps distinguish HS from G6PD deficiency (Type II pattern, corrected by glucose and ATP). The <b>EMA (Eosin-5-maleimide) binding test</b> by flow cytometry shows <b>decreased fluorescence</b> because EMA covalently binds Band 3 and Rh-associated proteins — their deficiency results in reduced binding. This is now considered the <b>best modern screening test</b> (sensitive, specific, and rapid). <b>SDS-PAGE (gel electrophoresis)</b> is the gold standard for identifying and quantifying specific membrane protein deficiencies. The <b>direct antiglobulin test (Coombs test) is negative</b>, confirming the non-immune nature of hemolysis and distinguishing HS from autoimmune hemolytic anemia (AIHA). Routine labs show elevated unconjugated bilirubin, elevated LDH, reduced haptoglobin, and <b>elevated MCHC</b>.<br><br><b>COMPLICATIONS</b><br>Hemolytic anemia with unconjugated jaundice is the primary manifestation. Pigment gallstones (calcium bilirubinate) form due to chronic hyperbilirubinemia. Splenomegaly is universal in symptomatic disease. <b>Aplastic crisis</b> is the most dangerous acute complication — caused by <b>Parvovirus B19</b> (infects and destroys erythroid precursors bearing the P antigen), leading to sudden cessation of RBC production and rapid worsening of anemia. <b>Megaloblastic crisis</b> occurs due to folate deficiency, as chronically accelerated erythropoiesis exhausts folate stores. <b>Hemolytic crisis</b> is triggered by infections causing transient worsening of hemolysis. Leg ulcers are rare but can occur due to microvascular sludging of rigid spherocytes.<br><br><b>ONE-LINERS FOR RAPID RECALL</b><br>Most common hereditary hemolytic anemia in Europeans → HS. Most common mutation → Ankyrin (ANK1). Most common inheritance → Autosomal Dominant. Fundamental defect → Vertical interaction failure → membrane vesiculation → reduced surface area. Hallmark routine lab → Elevated MCHC (>36 g/dL). Classic diagnostic test → Osmotic fragility test (increased). Best modern screening test → EMA binding (flow cytometry). Gold standard protein identification → SDS-PAGE. Coombs test → Negative. Site of hemolysis → Extravascular (splenic cords of Billroth). Aplastic crisis → Parvovirus B19. Megaloblastic crisis → Folate deficiency. Splenectomy → Curative for hemolysis, membrane defect persists. Gallstones → Pigment type (calcium bilirubinate). Protein 4.2 deficiency → AR, Japanese population. Credited to → Minkowski and Chauffard.
- Save the above text as
HS_anki.txt - Open Anki → File → Import
- Select the file
- Set Field separator to
| - Check "Allow HTML in fields"
- Map Field 1 → Front, Field 2 → Back
- Click Import