Cushing’s syndrome
Reading File
Reading File
Finding Sources
Searching PubMed
"Cushing syndrome"[MeSH Terms] AND diagnosis
Running Command
Reading File
Searching Images
Cushing syndrome clinical features moon face buffalo hump
Running Command
Reading File
Running Command
Reading File
Reading File
Running Command
Running Command
Running Command
Running Command
Running Command
Loading Image
Now I have all the material needed. Here is the comprehensive overview:
Cushing Syndrome
Based on Nelson Textbook of Pediatrics 2024 (Chapter 619) and Campbell-Walsh Urology
Definition
Cushing syndrome is the clinical state resulting from abnormally high blood levels of cortisol or other glucocorticoids, whether from exogenous administration or endogenous oversecretion. — Nelson 2024, Ch. 619
Etiology & Classification
Causes are divided into three main groups:
1. Exogenous (Iatrogenic) — Most Common Overall
The most frequent cause in the Western world. Synthetic glucocorticoids given orally, topically, or by inhalation can produce the syndrome, even at low doses. A careful history should inquire about herbal remedies, nasal sprays, and surreptitious use.
2. ACTH-Dependent (80–85% of endogenous cases)
| Cause | Key Facts |
|---|---|
| Cushing disease (pituitary corticotroph adenoma) | ~70% of endogenous CS; almost always a microadenoma <5 mm; ♀ > ♂ overall, but prepubertal males > females |
| Ectopic ACTH secretion | ~10% of endogenous CS; in children: associated with islet cell carcinoma of the pancreas, neuroblastoma, ganglioneuroblastoma, hemangiopericytoma, Wilms tumor, thymic carcinoid; causes the highest cortisol and most severe hypokalemia |
| Ectopic CRH secretion | Extremely rare |
3. ACTH-Independent (~15–20% of endogenous cases)
| Cause | Key Facts |
|---|---|
| Adrenal adenoma | Most common adrenal cause in children >7 yr |
| Adrenocortical carcinoma | ~8% of CS; poor prognosis; virilization often co-exists |
| PPNAD (Primary Pigmented Nodular Adrenocortical Disease) | <1%; normal-sized glands with black/brown nodules; associated with Carney complex (PRKAR1A gene, chr 17q22–24); classic paradoxical rise on high-dose dexamethasone suppression test |
| BMAH (Bilateral Macronodular Adrenal Hyperplasia) | <1%; ARMC5 tumor-suppressor gene abnormal in ~40% |
| McCune-Albright syndrome | GNAS (Gsα) somatic activating variant |
Pediatric note: In children under 5 years, adrenal causes (carcinoma, McCune-Albright) predominate over pituitary causes.
Pathophysiology
The hypothalamic-pituitary-adrenal (HPA) axis normally maintains cortisol within a circadian rhythm: peak at 8 AM, nadir at ~11 PM. CRH → ACTH → cortisol → negative feedback at hypothalamus and pituitary. In Cushing syndrome, this feedback loop is disrupted and the normal circadian rhythm is lost.
The adrenal cortex's zona fasciculata produces up to 20 mg cortisol/day under ACTH drive. Cortisol exerts effects on virtually every organ system (see figure below).
Clinical Manifestations
The most sensitive indicator of excess glucocorticoid secretion in children is growth failure, which generally precedes other signs. (Children with simple obesity are usually tall, whereas those with Cushing syndrome are short or have a decelerating growth rate.)
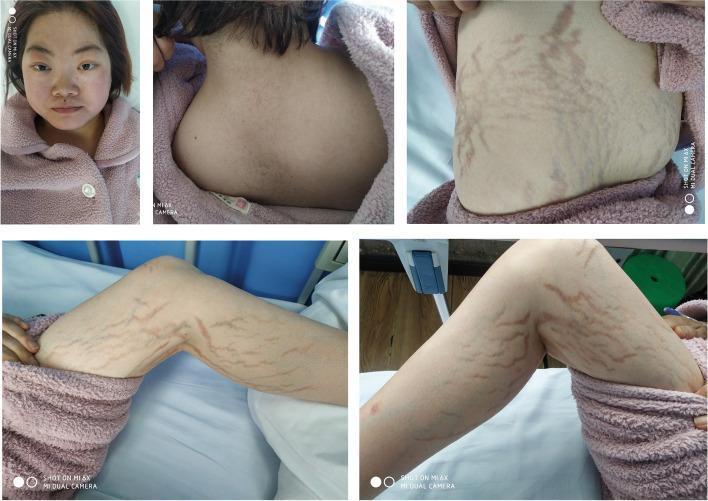
Table: Presenting Signs and Symptoms (Nelson 2024, Table 619.2)
| System | Features |
|---|---|
| Dermatologic | Moon facies (rounded, flushed), facial plethora, acne, easy bruising, supratemporal and supraclavicular fat pads, hirsutism, fine downy hair, violaceous striae (unusual in children <7 yr), fungal infections, acanthosis nigricans |
| Growth | Growth deceleration + weight gain; central obesity; "buffalo hump" (nonspecific) |
| Cardiovascular | Hypertension, coagulopathy |
| Metabolic | Hyperglycemia → frank diabetes, dyslipidemia, insulin resistance |
| Gonadal | Amenorrhea, virilization (clitoromegaly, pubic hair, deepened voice), gynecomastia; pubertal arrest or delay |
| Musculoskeletal | Osteoporosis, pathologic fractures, proximal muscle weakness |
| Neuropsychiatric | Depression, anxiety, mood swings, irritability, fatigue, headache |
| Other | Nephrolithiasis, increased susceptibility to infection |
In infants, generalized obesity is common and findings are more dramatic. In older children, truncal/facial obesity with relative limb sparing is typical.
Laboratory Findings & Diagnosis
Screening Tests (require ≥2 abnormal results to confirm)
| Test | Details |
|---|---|
| Late-night salivary cortisol | Circadian rhythm is lost in CS; midnight cortisol >4.4 μg/dL strongly suggests the diagnosis. Can be collected at home. |
| 24-hour urinary free cortisol (UFC) | Expressed per gram creatinine (corrects for body size and collection completeness). Elevated in CS. |
| Overnight dexamethasone suppression test (DST) | 25–30 μg/kg (max 2 mg) at 11 PM → 8 AM cortisol <5 μg/dL is normal. Failure to suppress = abnormal. Measure dexamethasone level to confirm adequate dosing. |
Distinguishing the Cause
Once CS is confirmed:
-
Measure plasma ACTH (two samples)
- ACTH <10 pg/mL → ACTH-independent → CT/MRI of adrenals
- ACTH >20 pg/mL → ACTH-dependent → pituitary MRI + further testing
- ACTH 10–20 pg/mL (indeterminate) → CRH stimulation test
-
High-dose dexamethasone suppression test (HDDSST): 30 μg/kg/24 hr × 2 days then 120 μg/kg/24 hr × 2 days. Suppression of cortisol with the larger dose (not smaller) = pituitary Cushing disease. ACTH-independent disease shows no suppression.
-
CRH stimulation: Patients with ACTH-dependent CS show an exaggerated ACTH and cortisol response; adrenal tumor patients show no response.
-
Bilateral Inferior Petrosal Sinus Sampling (BIPSS) after CRH stimulation: the gold standard to distinguish Cushing disease from ectopic ACTH. High petrosal:peripheral ACTH ratio confirms pituitary origin.
-
Imaging:
- CT detects virtually all adrenal tumors >1.5 cm
- Pituitary MRI (with gadolinium) for adenomas — but 50% of Cushing disease patients have no visible abnormality on MRI; and 10% of the general population have incidental pituitary findings
Algorithm summary (Fig. 619.2, Nelson 2024):
Confirmed CS → ACTH measurement
├── <10 pg/mL (ACTH-independent) → CT/MRI adrenals
│ └── Unilateral mass → adenoma/carcinoma
│ └── Bilateral → macronodular or micronodular hyperplasia
│
└── >20 pg/mL (ACTH-dependent) → Pituitary MRI + HDDSST + CRH test
├── Adenoma >6 mm + concordant tests → Surgery (Cushing disease confirmed)
├── Adenoma <6 mm or discordant tests → BIPSS
└── Negative pituitary imaging → CT/MRI neck/chest/abdomen/pelvis + octreoscan/PET → ectopic ACTH tumor
Differential Diagnosis
- Simple obesity: children are tall, cortisol suppresses normally with dexamethasone, nighttime salivary cortisol is normal
- Pseudo-Cushing states (may show false-positive cortisol tests): major depression, alcoholism, morbid obesity, poorly controlled diabetes
- Glucocorticoid resistance (Chrousos syndrome): elevated cortisol + ACTH without typical CS phenotype; presents with hypertension, hypokalemia, precocious pseudopuberty
- Exogenous steroid use (always exclude first with careful history)
Treatment
Cushing Disease (Pituitary)
- Transsphenoidal pituitary microsurgery is the treatment of choice in children
- Overall success rate: 60–98% (initial remission); long-term success 50–98%
- Low postoperative cortisol predicts long-term remission
- Relapse: reoperation or pituitary radiotherapy
- Pasireotide (somatostatin analog): approved for adults with persistent/recurrent disease post-surgery or surgical contraindication — inhibits ACTH secretion
- Cyproheptadine (serotonin antagonist): rarely used in children; remissions not sustained
Adrenal Causes
- Surgical resection (laparoscopic adrenalectomy where possible)
- For bilateral adrenalectomy: requires lifelong adrenal replacement therapy; risk of Nelson syndrome (~25% of adults post-bilateral adrenalectomy) — Nelson syndrome = markedly elevated ACTH with skin hyperpigmentation from unresected pituitary adenoma
Adrenal Steroidogenesis Inhibitors (preoperative or adjunct)
| Drug | Mechanism |
|---|---|
| Mitotane | Cytotoxic to adrenocortical cells; FDA-approved |
| Osilodrostat | Cortisol synthesis inhibitor; FDA-approved |
| Metyrapone | 11β-hydroxylase inhibitor |
| Ketoconazole / Levoketoconazole | Inhibits multiple steroidogenic enzymes; levoketoconazole in advanced development |
| Aminoglutethimide | Blocks cholesterol side-chain cleavage |
| Etomidate (IV) | For acute severe hypercortisolism |
| Mifepristone | Glucocorticoid receptor antagonist; used in limited cases |
Postoperative Management
- Replace cortisol 10 mg/m²/24 hr in three divided doses after immediate postoperative period — until HPA axis recovers (may take up to 30 months)
- Monitor for sepsis, pancreatitis, thrombosis, poor wound healing
- Expect catch-up growth and pubertal progress, but bone density remains abnormal and adult height is often compromised
- Check for GH deficiency post-treatment and treat if present
Genetics Summary (Fig. 619.1, Nelson 2024)
| Cause | Key Genes |
|---|---|
| Cushing disease | USP8, MEN1, CDKIs, CDKN1B/p27, AIP, DICER1 |
| PPNAD / Carney complex | PRKAR1A (chr 17q22–24), PDE11A, PDE8B, PRKACA |
| BMAH | ARMC5, MEN1, GNAS1, PDE11A, PDE8B |
| Adrenal adenoma | PRKACA (somatic activating variant), CTNNB1 |
| McCune-Albright | GNAS (somatic Gsα activating variant) |
| Ectopic ACTH | RET, MEN1 |
Recent Evidence (PubMed 2024–2025)
- A 2024 systematic review and meta-analysis (PMID 38888685) addressed the challenge of differentiating true Cushing syndrome from non-neoplastic hypercortisolism (pseudo-Cushing), highlighting the diagnostic conundrum in clinical practice.
- A 2025 meta-analysis on ectopic Cushing syndrome (PMID 40455890) characterized survival probabilities — ectopic CS carries a significantly worse prognosis than pituitary or adrenal forms.
- A 2024 meta-analysis (PMID 38840128) documented a spectrum of thyroid dysfunction in CS, including suppressed TSH and altered thyroid hormone metabolism.
Sources: Nelson Textbook of Pediatrics 2024, Ch. 619 (attached PDF) | Campbell-Walsh-Wein Urology, Ch. 106
This is a shared conversation. Sign in to Orris to start your own chat.