I want you to create a summary of the pathology aspect of hemolytic uremic syndrome, covering almost 100% topics of pathology-related aspect which can be asked for NEET PG 2026 clinical MCQs.
hemolytic uremic syndrome renal biopsy thrombotic microangiopathy pathology
Hemolytic Uremic Syndrome (HUS) — Complete Pathology Summary for NEET PG 2026
1. DEFINITION & CLASSIFICATION
- Microangiopathic Hemolytic Anemia (MAHA)
- Thrombocytopenia
- Acute Kidney Injury (AKI) — the dominant organ involved in HUS
TMA is a clinicopathological syndrome: thrombocytopenia + MAHA evidenced by schistocytes on peripheral smear + elevated LDH + reticulocytosis + suppressed haptoglobin → microvascular occlusion → multi-organ dysfunction. — National Kidney Foundation Primer on Kidney Diseases, 8e
Classification of HUS (NEET High-Yield Table)
| Type | Also Called | Cause | Age |
|---|---|---|---|
| Typical HUS (90% of childhood HUS) | STEC-HUS / D+ HUS | Shiga toxin–producing E. coli (esp. O157:H7) or Shigella dysenteriae type 1 | Children < 5 yrs |
| Atypical HUS (aHUS) | D− HUS / complement-mediated HUS | Genetic/autoimmune complement dysregulation (alternative pathway) | Any age, but ~67% in childhood |
| Secondary HUS | — | Drugs, malignancy, pregnancy, transplant, HIV | Adults |
2. PATHOGENESIS OF TYPICAL (STEC) HUS
Step-by-step mechanism (MCQ favorite):
- Ingestion of STEC (most commonly E. coli O157:H7; also O104:H4, O26, O111 etc.) via undercooked beef, unpasteurized dairy, contaminated vegetables/water
- STEC colonizes intestinal mucosa via intimin (protein encoded by eaeA gene) → attaching/effacing lesion
- Shiga toxin (Stx, verotoxin) is produced — Stx2 is more nephrotoxic than Stx1
- Toxin crosses the intestinal epithelium → enters circulation attached to leukocytes, erythrocytes, and platelets
- Stx binds to globotriaosylceramide (Gb3) receptors on endothelial cells via its pentameric B subunit (via clathrin-coated pits → retrograde transport via Golgi to ER)
- A subunit inhibits ribosomal function (28S rRNA) → protein synthesis inhibition → endothelial cell death
- Why kidney is most affected: Renal glomerular endothelial cells express the highest density of Gb3; also podocytes, mesangial cells, and tubular epithelium express Gb3
- Why children are more affected: Higher Gb3 expression on renal endothelium in children; adults have circulating anti-Stx antibodies
- Cytokine amplification: Stx releases IL-1, TNF-α, IL-6 → procoagulant milieu
- Low-dose Stx: activates endothelial cells → leukocyte adhesion, ↑endothelin, ↓nitric oxide, platelet adhesion
- High-dose Stx: endothelial cell death → microvascular thrombosis in glomerular capillaries, afferent arterioles, and interlobular arteries
"Renal glomerular endothelial cells are especially vulnerable because they express the membrane receptor for Shiga toxin." — Robbins Basic Pathology
Role of Complement in STEC-HUS:
- Low C3 levels in up to 50% of STEC-HUS cases
- Stx induces P-selectin expression → increases alternative pathway (AP) complement activation
- Rare co-existing complement mutations → worse prognosis
3. PATHOGENESIS OF ATYPICAL HUS (aHUS)
Core defect: Dysregulation of the Alternative Complement Pathway
| Mutation | Protein affected | Frequency | Prognosis |
|---|---|---|---|
| CFH (most common) | Factor H (key downregulator of AP; degrades C3b) | Most common | Death/ESRD >50%; recurrence >75% |
| CFI | Factor I (cleaves C3b and C4b) | Common | Death/ESRD >50%; recurrence >75% |
| MCP (CD46) | Membrane cofactor protein (cofactor for CFI) | — | Better prognosis; recurrence in transplant |
| C3 | C3 itself (gain of function) | — | Poor |
| CFB | Factor B (component of C3bBb convertase; gain of function) | — | Death/ESRD >50% |
| Thrombomodulin | Thrombomodulin | — | Death/ESRD >50% |
| Anti-CFH antibodies | Autoimmune | ~6% of aHUS | "Autoimmune HUS"; mostly < 16 years |
4. KEY PATHOLOGICAL LESIONS (HISTOPATHOLOGY)
🔬 Light Microscopy (LM) — Hallmark features of TMA in HUS:
| Feature | Details |
|---|---|
| Microthrombi | In glomerular capillaries, afferent arterioles, interlobular arteries |
| Endothelial swelling | Endothelial cell detachment from GBM |
| Widening of subendothelial space | Accumulation of fluffy material |
| GBM duplication/splitting ("double contour") | Due to new basement membrane formation |
| Mesangiolysis | Dissolution/lysis of mesangial cells + loose mesangial matrix = mesangial expansion |
| Glomerular capillary congestion | Capillary lumen narrowing/occlusion |
| Intimal thickening | In arterioles in idiopathic/familial forms |
| "Onion-skin" lesion | Concentric myointimal hyperplasia of arterioles (more in aHUS/chronic) |
| Cortical necrosis | In severe cases (patchy) |
| Crescent formation | Uncommon |
| FSGS | Late sequela after acute HUS (especially in children with persistent hypertension) |
In STEC-HUS / young children: glomerular injury predominates In idiopathic/familial/adult forms: arterial and arteriolar injury predominates → secondary glomerular ischemia with retraction of glomerular tuft — Comprehensive Clinical Nephrology, 7e
EM (Electron Microscopy) findings:
- Swelling and detachment of endothelial cells from the basement membrane
- Accumulation of fluffy electron-lucent material in the subendothelium (widened subendothelial space)
- Intraluminal platelet thrombi
- Partial or complete obstruction of vessel lumina
- EM changes are similar to: scleroderma, malignant nephrosclerosis, chronic transplant rejection, calcineurin inhibitor nephrotoxicity
Immunofluorescence (IF):
- Negative for immunoglobulins (IgG, IgA, IgM) — KEY distinguishing feature
- Variable fibrinogen deposits in glomeruli and arterioles
- Some C3 staining may be present (especially in aHUS)
- No immune complex deposition (unlike MPGN, IgAN, etc.)
"Except for varying amounts of fibrinogen in the glomeruli and arterioles, immunofluorescence studies are typically negative for immunoglobulins and complement." — Robbins Basic Pathology
Kidney biopsy summary:
- Microthrombi in glomerular capillaries + arterioles
- Mesangiolysis (mesangial expansion with loose material)
- Endothelial swelling
- Double contour formation (GBM)
- IF: negative for Ig, some fibrinogen and C3
- Difficult to differentiate various TMA causes on pathology alone
5. PERIPHERAL BLOOD SMEAR — The Diagnostic Hallmark
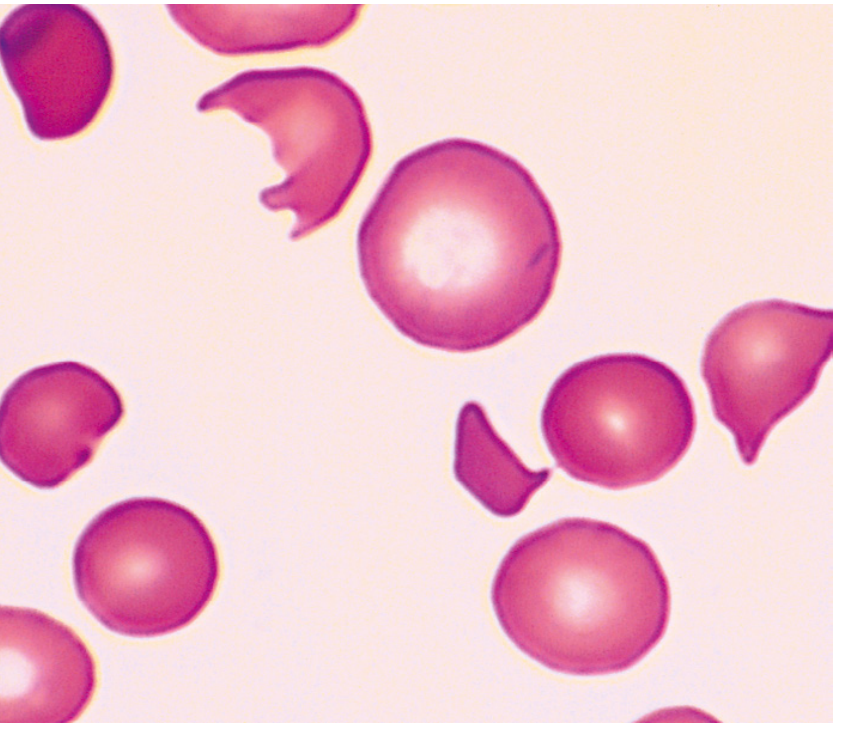
| Cell Type | Description |
|---|---|
| Schistocytes | Fragmented red cells — hallmark of MAHA |
| Helmet cells | Schistocyte variant |
| Burr cells (echinocytes) | Schistocyte variant |
| Triangle cells | Schistocyte variant |
6. RENAL BIOPSY HISTOPATHOLOGY IMAGE
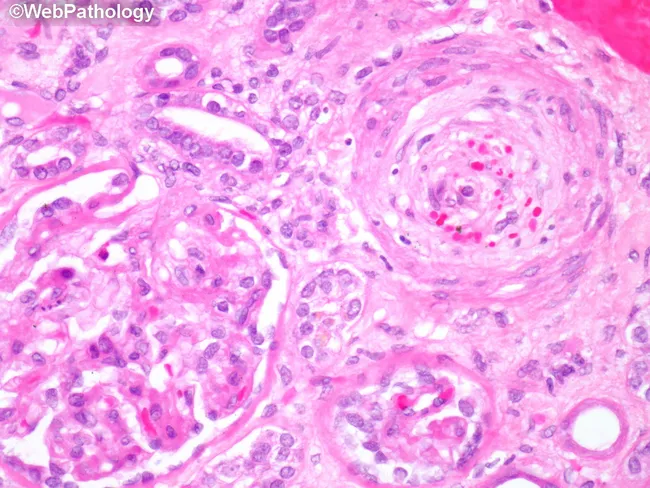
7. LABORATORY FINDINGS (Pathology of Investigation)
| Investigation | Finding in HUS | Significance |
|---|---|---|
| Peripheral smear | Schistocytes (fragmented RBCs) | Microangiopathic hemolysis |
| Hemoglobin | ↓ (moderate-severe anemia) | Hemolysis |
| LDH | ↑↑↑ | Hemolysis + tissue injury |
| Haptoglobin | ↓ (suppressed) | Intravascular hemolysis |
| Reticulocyte count | ↑ | Compensatory erythropoiesis |
| Indirect bilirubin | ↑ (usually mild) | Hemolysis |
| Direct bilirubin | Normal | Not biliary |
| Direct Coombs test | NEGATIVE | Non-immune hemolysis (key MCQ!) |
| Platelets | ↓ (<100,000/μL) | Consumption in microthrombi |
| PT/aPTT | Normal | KEY: distinguishes from DIC |
| Creatinine | ↑ (subacute worsening) | AKI |
| Urinalysis | Hematuria, proteinuria, granular casts | Glomerular injury |
| ADAMTS13 | Normal (>10% activity) | Distinguishes from TTP |
| Stool culture | STEC positive (O157:H7) | Confirms typical HUS |
| C3 levels | Low in ~50% of STEC-HUS; persistently low = poor prognosis | Complement activation |
| CFH/CFI/MCP mutations | In aHUS | Genetic workup |
8. HUS vs TTP vs DIC — High-Yield Comparison Table
| Feature | HUS | TTP | DIC |
|---|---|---|---|
| Age | Children (STEC) | Adults | Any |
| Trigger | E. coli O157:H7 | Autoimmune/ADAMTS13↓ | Sepsis, trauma, malignancy |
| Dominant organ | Kidney (AKI) | Brain (neuro signs) | Multi-organ |
| Fever | Absent (key!) | Present (classic pentad) | May be present |
| Schistocytes | Present | Present (>3/HPF) | Present |
| Schistocyte count | Less | More (>3/HPF) | Variable |
| ADAMTS13 | Normal | <10% (severe deficiency) | Normal |
| PT/aPTT | Normal | Normal | Prolonged |
| Fibrinogen | Normal | Normal | Decreased |
| D-dimers | Normal/mild ↑ | Normal | Elevated |
| Microthrombi | Kidney >> brain | Brain > kidney | Widespread |
| Coombs test | Negative | Negative | Negative |
| STEC | Positive | Negative | Negative |
| Treatment | Supportive; eculizumab (aHUS) | Plasmapheresis | Treat underlying cause |
9. TISSUE DISTRIBUTION OF MICROTHROMBI
| Organ | HUS | TTP |
|---|---|---|
| Kidney | Primary target | Present but less dominant |
| Brain | Less common (<50%) | Primary target |
| Pancreas | Can occur | Present |
| Heart | Rare | Present |
| Adrenals | Rare | Present |
| Skin | Petechiae/retiform purpura | Petechiae |
10. MECHANISM OF MICROANGIOPATHIC HEMOLYSIS
- HUS
- TTP
- DIC
- Malignant hypertension
- SLE
- Preeclampsia/HELLP
- Disseminated cancer
- Prosthetic cardiac valves
- Scleroderma (scleroderma renal crisis)
- Antiphospholipid antibody syndrome
- Drug-induced TMA (mitomycin C, gemcitabine, cyclosporin, quinine, valacyclovir)
11. COMPLEMENT PATHWAY — HIGH-YIELD NEET POINTS
Alternative Pathway C3 Convertase = C3bBb
- C3b + Factor B (cleaved by Factor D) → C3bBb (alternative pathway C3 convertase)
- C3bBb + properdin → stabilized convertase → C5 convertase → C5a + C5b → MAC (C5b-9)
- ~80% of all complement activity flows through alternative pathway amplification loop
Regulators lost in aHUS:
| Regulator | Function | Gene |
|---|---|---|
| Factor H (CFH) | Degrades C3b; most important plasma regulator | CFH |
| Factor I (CFI) | Cleaves C3b and C4b (requires cofactors) | CFI |
| MCP (CD46) | Cofactor for Factor I on cell surfaces | CD46 |
| Thrombomodulin | Activates Factor I; inactivates C3a and C5a | THBD |
Activating mutations (gain-of-function) in aHUS:
- C3 — gain of function → more C3b generated
- Factor B (CFB) — gain of function → more stable C3bBb convertase
12. DRUGS CAUSING SECONDARY HUS/TMA
| Drug | Association |
|---|---|
| Mitomycin C | Most common chemotherapy cause (dose-dependent) |
| Gemcitabine | Not dose-dependent; gastric, lung, colorectal, pancreatic, breast ca |
| Cyclosporine/Tacrolimus (calcineurin inhibitors) | Post-transplant TMA |
| Quinine | Immune-mediated |
| Oral contraceptives | Trigger in CFH (8%) and CFI (20%) mutations |
| VEGF inhibitors (bevacizumab) | TMA through endothelial injury |
| Cisplatin, bleomycin | Rare |
| Valacyclovir (high dose) | In HIV/immunocompromised patients |
| Cocaine | Secondary TMA |
HUS onset after chemotherapy: typically 4–8 weeks after last dose, but can be months later. — Harrison's 22e
13. PNEUMOCOCCAL HUS
- Streptococcus pneumoniae infection can precipitate HUS
- Mechanism: Neuraminidase produced by pneumococcus cleaves sialic acid from RBCs, platelets, and glomerular endothelium → exposes Thomsen-Friedenreich (T) antigen
- Naturally occurring IgM anti-T antibodies bind → hemolysis + endothelial injury
- Key MCQ: Avoid plasma exchange (FFP contains anti-T antibodies → worsens condition)
- Treatment: Antibiotics + supportive care; no FFP
14. PREGNANCY-ASSOCIATED HUS
- 20% of aHUS cases are triggered by pregnancy
- Typically presents postpartum (most common timing — see differential diagnosis figure from Brenner's)
- High proportion have underlying complement mutations (especially CFH, CFI)
- TTP: more common in 2nd and 3rd trimester
- HELLP: complicates 10–20% of preeclampsia cases; ~0.1% of all pregnancies
- Eculizumab is safe in pregnancy for aHUS/PNH
15. TREATMENT — PATHOLOGY IMPLICATIONS
| Treatment | Indication | Mechanism |
|---|---|---|
| Supportive care (dialysis, fluid, BP control) | STEC-HUS | Self-limiting; avoid antibiotics (accelerate Stx release) |
| Plasma Exchange (PEX) | TTP (life-saving); aHUS (less effective) | Removes ADAMTS13 antibodies; replaces ADAMTS13 |
| Eculizumab | aHUS (first-line, FDA-approved); refractory TTP | Humanized monoclonal antibody → binds C5 → blocks C5a + C5b → prevents MAC formation |
| Ravulizumab | aHUS (extended half-life version of eculizumab) | Same mechanism; blocks terminal complement at C5 |
| Rituximab | Refractory TTP; autoimmune aHUS; chemotherapy-induced HUS | Anti-CD20; depletes B cells → reduces autoantibodies |
| Caplacizumab | TTP | Anti-VWF nanobody; blocks vWF-platelet interaction |
| Vaccination | Before eculizumab/ravulizumab | Mandatory: Neisseria meningitidis vaccination (terminal complement blocked → meningococcal risk) |
Antibiotics in STEC-HUS = CONTRAINDICATED → accelerate Shiga toxin release → worsen HUS
16. PROGNOSIS — PATHOLOGY CORRELATIONS
| Variable | Significance |
|---|---|
| CFH, CFI, CFB, thrombomodulin mutations | Death or ESRD in >50% |
| MCP (CD46) mutations | Better prognosis; high recurrence after transplant |
| CFH/CFI mutations | Recurrence in >75% after transplant |
| Anti-CFH antibodies | Good response to immunosuppression |
| STEC-HUS with acute dialysis | Most recover in weeks |
| Long-term STEC-HUS (15–25 yrs) | Not uniformly favorable |
| Persistent low C3 in STEC-HUS | Poorer prognosis |
| Cortical necrosis on biopsy | Severe; may not recover |
| FSGS as late sequela | Chronic hypertension + progressive CKD |
17. KEY MCQ ANCHOR FACTS (MUST-REMEMBER)
| Fact | Answer |
|---|---|
| Most common cause of AKI requiring dialysis in children 1–5 years | HUS |
| Receptor for Shiga toxin on endothelium | Gb3 (globotriaosylceramide) |
| Most nephrotoxic Shiga toxin | Stx2 |
| Active unit of Shiga toxin causing cell death | A subunit (inhibits ribosome) |
| Binding unit of Shiga toxin | B subunit (pentameric) |
| Most common STEC serotype causing HUS in Europe/North America | E. coli O157:H7 |
| Largest recorded HUS outbreak (2011, Germany) | E. coli O104:H4 (fenugreek sprouts) |
| HUS in Asia/Africa (Shigella cause) | Shigella dysenteriae serotype 1 |
| Dominant feature in HUS (vs TTP) | Renal failure (AKI) |
| Dominant feature in TTP (vs HUS) | Neurological signs |
| Coombs test in HUS | Negative (non-immune hemolysis) |
| PT/aPTT in HUS | Normal (unlike DIC) |
| ADAMTS13 in HUS | Normal (unlike TTP <10%) |
| IF on renal biopsy in HUS | Negative for Ig; ± fibrinogen/C3 |
| EM hallmark of TMA | Endothelial swelling + subendothelial fluffy material + intraluminal thrombi |
| Most important complement regulator mutated in aHUS | Factor H (CFH) |
| Eculizumab target | C5 |
| Mandatory vaccination before eculizumab | N. meningitidis |
| Why avoid antibiotics in STEC-HUS | Accelerate Shiga toxin release |
| Why avoid FFP in pneumococcal HUS | FFP contains anti-T antibodies → worsens disease |
| Chemotherapy most commonly causing HUS | Mitomycin C |
| % of childhood HUS due to STEC | >90% |
| % of STEC infections that progress to HUS | 5–10% |
| Oral contraceptives trigger aHUS in CFI mutations | 20% (CFH = 8%) |
| Schistocytes: more in TTP or HUS? | More in TTP (>3/HPF) |
| Microthrombi in HUS (location) | Glomerular capillaries + afferent arterioles |
| Microthrombi in TTP (location) | Pancreas, heart, adrenals, brain |
| TMA pathology differentiable from non-TMA causes on biopsy? | No — cannot differentiate TMA causes by pathology alone |
| "Double contour" (tram-track) GBM in HUS | Due to new BM formation over widened subendothelial space |
| Mesangiolysis | Lysis/dissolution of mesangial cells + loosening of mesangial matrix — seen in HUS |
| aHUS trigger: pregnancy — typical timing | Postpartum |
| % of aHUS triggered by pregnancy | 20% |
| Gene mutation giving best prognosis in aHUS | MCP (CD46) |
| DGKE mutation HUS | Diacylglycerol kinase ε — non-complement, eculizumab NON-responsive |
| MMACHC mutation HUS | Methylmalonic aciduria + homocystinuria type C — eculizumab NON-responsive |
| C5 polymorphism p.R885H | Non-responsive to eculizumab |
18. PATHOLOGICAL DIFFERENTIAL DIAGNOSIS OF TMA
- STEC-HUS
- aHUS (complement-mediated)
- TTP (ADAMTS13 deficiency)
- Malignant hypertension
- Scleroderma renal crisis
- Antiphospholipid antibody syndrome (APS)
- SLE-associated TMA
- Calcineurin inhibitor (cyclosporine/tacrolimus) nephrotoxicity
- Chronic transplant rejection
- Drug-induced TMA (mitomycin C, gemcitabine)
- Preeclampsia/HELLP
- HIV-associated TMA
- Pregnancy-associated TMA
- Bone marrow transplant–associated TMA
Clinical context + laboratory findings (ADAMTS13, STEC culture, complement levels, genetic testing) are essential to distinguish these conditions.
Now I want you to prepare a summary in the form of an Anki card to be imported to AnkiDroid app. It should only be one card. The front should be Hemolytic Uremic Syndrome. The back should be the summary with proper spacing, punctuation, so that it can be easily readable. It should only include the pathology aspects and only the important points that are required for the exam. But it should be a self-explanatory summary. Like I should be able to understand the mechanism without mugging it up. The explanation should be present, but only for what is required. And it should cover almost 100% of the as part of Hemolytic Uremic Syndrome pathology that can be asked for the exam. It should be in a text form where I can copy and paste to a text app and then import to AnkiDroid app.
#separator:tab
#html:false
#notetype:Basic
Hemolytic Uremic Syndrome (Pathology — NEET PG 2026) HEMOLYTIC UREMIC SYNDROME — PATHOLOGY SUMMARY
=== DEFINITION / TRIAD ===
HUS = Thrombotic Microangiopathy (TMA) with:
1. Microangiopathic Hemolytic Anemia (MAHA)
2. Thrombocytopenia
3. Acute Kidney Injury (AKI) — dominant organ in HUS
=== CLASSIFICATION ===
Typical HUS (D+ HUS / STEC-HUS):
- Cause: Shiga toxin-producing E. coli (E. coli O157:H7 — most common in Europe/North America) or Shigella dysenteriae type 1 (Asia/Africa)
- Age: Children <5 years
- Diarrhea prodrome (bloody in ~60%)
- Accounts for >90% of childhood HUS
Atypical HUS (D− HUS / aHUS):
- Cause: Genetic or autoimmune dysregulation of alternative complement pathway
- Any age; ~67% in childhood
- No diarrhea prodrome
Secondary HUS:
- Drugs, malignancy, pregnancy, HIV, transplant
=== PATHOGENESIS OF TYPICAL HUS ===
How Shiga toxin causes kidney injury:
1. STEC colonizes gut via intimin (eaeA gene) — attaching/effacing lesion
2. Shiga toxin (Stx) produced — Stx2 more nephrotoxic than Stx1
3. Toxin enters circulation attached to leukocytes, RBCs, platelets
4. B subunit (pentameric) binds Gb3 (globotriaosylceramide) receptor on renal endothelium — internalized via clathrin-coated pits → retrograde transport (Golgi → ER)
5. A subunit inhibits ribosomal protein synthesis → endothelial cell death
6. Result: endothelial injury → platelet activation → microvascular thrombosis
Why kidney is most affected:
- Glomerular endothelium expresses highest density of Gb3
- Also on podocytes, mesangial cells, tubular epithelium
Why children more affected:
- Higher Gb3 expression on renal endothelium
- Adults have protective anti-Stx antibodies
Additional pathogenic mechanisms:
- Low-dose Stx: activates endothelium → increased endothelin, decreased NO, leukocyte adhesion (vasoconstriction + platelet adhesion)
- High-dose Stx: endothelial cell death
- Stx releases IL-1, TNF-alpha, IL-6 → amplifies procoagulant milieu
- Complement (AP) activated in ~50% STEC-HUS (low C3); persistent low C3 = poor prognosis
=== PATHOGENESIS OF ATYPICAL HUS ===
Core defect: Uncontrolled activation of alternative complement pathway C3 convertase (C3bBb)
Normal protection: Factor H (CFH), Factor I (CFI), MCP (CD46), Thrombomodulin → degrade C3b → prevent runaway complement
Loss-of-function mutations (allow unopposed C3b/complement activation):
- CFH (most common) — key downregulator; heterozygotes → abnormal CFH inactivates normal CFH
- CFI — cleaves C3b and C4b
- MCP / CD46 — cell-surface cofactor for CFI (best prognosis; high transplant recurrence)
- Thrombomodulin
Gain-of-function mutations (increase convertase activity):
- C3 — more C3b generated
- Factor B (CFB) — more stable C3bBb convertase
Death/ESRD >50% and recurrence >75%: CFH, CFI, CFB, thrombomodulin mutations
Best prognosis: MCP mutations
Autoimmune aHUS: anti-CFH antibodies (~6% of aHUS; mostly <16 years)
Triggers in carriers: Pregnancy (postpartum; 20% of aHUS), oral contraceptives (CFH: 8%; CFI: 20%), infections
Eculizumab NON-responsive aHUS: DGKE, MMACHC, INF2 mutations; C5 polymorphism p.R885H
=== RENAL HISTOPATHOLOGY (BIOPSY FINDINGS) ===
Light Microscopy:
- Microthrombi in glomerular capillaries, afferent arterioles, interlobular arteries
- Endothelial swelling and detachment from GBM
- Widening of subendothelial space (fluffy material)
- GBM duplication / "double contour" (tram-track) — new BM forms over injured zone
- Mesangiolysis — lysis of mesangial cells + loose mesangial matrix
- "Onion-skin" arteriolar lesion — concentric myointimal hyperplasia (more in aHUS/chronic)
- Cortical necrosis — patchy, in severe cases
- Crescent formation — uncommon
- FSGS — late sequela (chronic hypertension + progressive CKD)
Pattern distinction:
- STEC-HUS / young children: glomerular injury predominates
- Idiopathic/familial/adult aHUS: arteriolar injury predominates → secondary glomerular ischemia
Electron Microscopy:
- Endothelial cell swelling and detachment from BM
- Subendothelial fluffy electron-lucent material (widened subendothelial space)
- Intraluminal platelet thrombi
- Similar appearance to: scleroderma, malignant nephrosclerosis, chronic transplant rejection, calcineurin inhibitor nephrotoxicity
Immunofluorescence:
- NEGATIVE for immunoglobulins (IgG, IgA, IgM) — KEY distinguishing point
- Variable fibrinogen in glomeruli and arterioles
- Some C3 staining may be present (esp. aHUS)
- No immune complex deposition
- Cannot differentiate causes of TMA by pathology alone — clinical/lab context required
=== PERIPHERAL BLOOD SMEAR ===
Schistocytes (fragmented RBCs) = hallmark of MAHA
Morphology: helmet cells, burr cells, triangle cells
Mechanism: microvascular thrombi → shear stress on passing RBCs → mechanical fragmentation → intravascular hemolysis
Schistocyte count: more numerous in TTP (>3/HPF) than HUS
=== LABORATORY PATHOLOGY ===
- Hemoglobin: decreased (hemolysis)
- LDH: markedly elevated (hemolysis + tissue injury)
- Haptoglobin: decreased/absent (intravascular hemolysis)
- Indirect bilirubin: mildly elevated
- Reticulocyte count: elevated
- Direct Coombs test: NEGATIVE (non-immune, mechanical hemolysis)
- Platelets: <100,000/uL (consumption in thrombi)
- PT/aPTT: NORMAL (key — differentiates from DIC)
- Fibrinogen: Normal (key — differentiates from DIC)
- Creatinine: elevated, subacutely worsening
- Urinalysis: hematuria, proteinuria, granular casts
- ADAMTS13: NORMAL (key — differentiates from TTP, where <10%)
- C3: low in ~50% STEC-HUS; persistent low = poor prognosis
- Stool culture: STEC positive in typical HUS
=== HUS vs TTP vs DIC ===
Feature | HUS | TTP | DIC
Dominant organ | Kidney (AKI) | Brain (neuro) | Multi-organ
Fever | Absent | Present | Variable
Schistocytes | Present | Present (>3/HPF) | Present
ADAMTS13 | Normal | <10% (deficient) | Normal
PT/aPTT | Normal | Normal | Prolonged
Fibrinogen | Normal | Normal | Decreased
D-dimers | Normal/mild up | Normal | Elevated
Treatment | Supportive/eculizumab | Plasmapheresis | Treat cause
=== MICROTHROMBI DISTRIBUTION ===
HUS: primarily kidneys (glomerular capillaries + afferent arterioles)
TTP: kidneys, pancreas, heart, adrenals, brain (more widespread)
Neurological involvement in HUS: <50% (less common)
=== MECHANISM OF MAHA (MACROANSWER) ===
Any cause of luminal narrowing (thrombus, fibrin) in microvasculature → shear stress on RBCs → fragmentation → schistocytes + intravascular hemolysis
Other causes of MAHA (MCQ list): TTP, DIC, malignant hypertension, SLE, preeclampsia/HELLP, disseminated cancer, prosthetic cardiac valves, scleroderma renal crisis, APS, drugs (mitomycin C, gemcitabine)
=== COMPLEMENT CASCADE — HIGH-YIELD ===
Alternative pathway C3 convertase = C3bBb (C3b + Factor B, cleaved by Factor D)
AP amplification = ~80% of all complement activity
Terminal pathway: C5 convertase → C5a (anaphylatoxin) + C5b → C5b-9 = MAC (membrane attack complex)
Eculizumab: binds C5 → blocks C5a + C5b → prevents MAC → stops endothelial/platelet damage
Ravulizumab: same mechanism as eculizumab, extended half-life, FDA approved for aHUS
=== DRUGS CAUSING SECONDARY HUS ===
- Mitomycin C: most common chemotherapy cause; dose-dependent; onset 4-8 weeks after last dose
- Gemcitabine: NOT dose-dependent; gastric/lung/colorectal/pancreatic/breast cancers
- Cyclosporine/Tacrolimus: post-transplant TMA
- Quinine: immune-mediated
- VEGF inhibitors (bevacizumab): endothelial injury → TMA
- Valacyclovir (high dose in HIV): rare but fatal
- Oral contraceptives: trigger aHUS in complement mutation carriers
=== PNEUMOCOCCAL HUS ===
- Streptococcus pneumoniae → neuraminidase cleaves sialic acid from RBCs, platelets, endothelium → exposes Thomsen-Friedenreich (T) antigen
- Natural IgM anti-T antibodies bind → hemolysis + endothelial injury
- KEY: AVOID plasma exchange / FFP (FFP contains anti-T antibodies → worsens disease)
- Treatment: antibiotics + supportive care only
=== PROGNOSIS — PATHOLOGICAL CORRELATES ===
- CFH, CFI, CFB, thrombomodulin mutations: Death/ESRD >50%; recurrence >75%
- MCP (CD46) mutations: best prognosis; high recurrence in transplant
- Cortical necrosis on biopsy: severe; may not recover
- FSGS as late sequela: chronic CKD
- STEC-HUS with dialysis: most recover in weeks; long-term (15-25 yrs) not uniformly favorable
- Anti-CFH antibodies: good response to immunosuppression; diagnosed mostly <16 yrs
=== TREATMENT — KEY POINTS ===
- STEC-HUS: supportive only — NO antibiotics (accelerate Stx release), NO plasma exchange (not effective)
- aHUS: Eculizumab (first-line, FDA-approved) — blocks C5; mandatory N. meningitidis vaccination before starting
- TTP: Plasma exchange (life-saving) — removes anti-ADAMTS13 antibodies + replaces ADAMTS13
- Rituximab: refractory TTP, autoimmune aHUS, chemotherapy-induced HUS
- Caplacizumab: TTP — blocks vWF-platelet interaction
- Pneumococcal HUS: NO FFP
=== MUST-KNOW MCQ ANCHORS ===
- Most common cause of AKI requiring dialysis in children 1-5 years = HUS
- Shiga toxin receptor on endothelium = Gb3 (globotriaosylceramide)
- More nephrotoxic Stx = Stx2
- Stx A subunit = inhibits ribosome (protein synthesis); B subunit = binds Gb3
- Most common STEC serotype (Europe/N. America) = E. coli O157:H7
- Largest HUS outbreak (2011, Germany) = E. coli O104:H4 (fenugreek sprouts)
- Shiga HUS in Asia/Africa = Shigella dysenteriae type 1
- Coombs test in HUS = NEGATIVE
- IF on renal biopsy = negative for Ig (± fibrinogen/C3)
- EM hallmark = subendothelial fluffy material + endothelial swelling + intraluminal thrombi
- Cannot differentiate TMA causes on biopsy alone
- Most important complement regulator mutated in aHUS = Factor H (CFH)
- Eculizumab target = C5
- Mandatory pre-eculizumab vaccination = N. meningitidis
- Avoid antibiotics in STEC-HUS = accelerate Stx release
- Avoid FFP in pneumococcal HUS = contains anti-T antibodies
- Most common chemotherapy causing HUS = Mitomycin C
- % STEC progressing to HUS = 5-10%
- Oral contraceptives trigger aHUS: CFH = 8%, CFI = 20%
- Pregnancy-associated aHUS timing = postpartum (20% of all aHUS)
- ADAMTS13 inhibited by Stx = yes (Stx inhibits endothelial ADAMTS13 production)
- Eculizumab non-responsive mutations = DGKE, MMACHC, INF2, C5 p.R885H
- Copy everything above (from
#separator:tabto the last line) - Paste into a plain text editor (Notepad, Google Keep, etc.) and save as
HUS_Pathology.txt - In AnkiDroid → Menu → Import → select the
.txtfile - Confirm separator is Tab and notetype is Basic
- Import — one card will be created with the front/back as set
The#html:falseheader tells Anki to render it as plain text (no HTML parsing), so the spacing and formatting you see above will display cleanly on the card back.
How does that get look? The front card is the summary and the back card is the heading. I wanted them to both, the front should be the heading and the back should be the summary.
#separator:tab
#html:false
#notetype:Basic
Hemolytic Uremic Syndrome — Pathology (NEET PG 2026) HEMOLYTIC UREMIC SYNDROME — PATHOLOGY SUMMARY
=== DEFINITION / TRIAD ===
HUS = Thrombotic Microangiopathy (TMA) with:
1. Microangiopathic Hemolytic Anemia (MAHA)
2. Thrombocytopenia
3. Acute Kidney Injury (AKI) — dominant organ in HUS
=== CLASSIFICATION ===
Typical HUS (D+ HUS / STEC-HUS):
- Cause: Shiga toxin-producing E. coli (O157:H7 — most common in Europe/N. America) or Shigella dysenteriae type 1 (Asia/Africa)
- Age: Children <5 years; diarrhea (bloody in ~60%) prodrome
- >90% of childhood HUS
Atypical HUS (D- HUS / aHUS):
- Cause: Genetic or autoimmune dysregulation of alternative complement pathway
- Any age; ~67% in childhood; NO diarrhea prodrome
Secondary HUS: Drugs, malignancy, pregnancy, HIV, transplant
=== PATHOGENESIS OF TYPICAL HUS ===
How Shiga toxin causes kidney injury:
1. STEC colonizes gut via intimin (eaeA gene) — attaching/effacing lesion
2. Shiga toxin (Stx) produced — Stx2 is more nephrotoxic than Stx1
3. Toxin enters circulation attached to leukocytes, RBCs, platelets
4. B subunit (pentameric) binds Gb3 (globotriaosylceramide) receptor on renal endothelium — internalized via clathrin-coated pits → retrograde transport via Golgi → ER
5. A subunit inhibits ribosomal protein synthesis → endothelial cell death
6. Result: endothelial injury → platelet activation → microvascular thrombosis
Why kidney is most affected:
- Glomerular endothelium expresses highest density of Gb3
- Also expressed on podocytes, mesangial cells, tubular epithelium
Why children more affected:
- Higher Gb3 expression on renal endothelium in children
- Adults have protective circulating anti-Stx antibodies
Additional mechanisms:
- Low-dose Stx: activates endothelium → increased endothelin, decreased NO → vasoconstriction + platelet adhesion
- High-dose Stx: direct endothelial cell death
- Stx releases IL-1, TNF-alpha, IL-6 → amplifies procoagulant milieu
- Complement (AP) activated in ~50% of STEC-HUS (low C3); persistent low C3 = poor prognosis
=== PATHOGENESIS OF ATYPICAL HUS ===
Core defect: Uncontrolled activation of alternative complement pathway C3 convertase (C3bBb)
Normal regulation: Factor H (CFH), Factor I (CFI), MCP (CD46), Thrombomodulin → degrade C3b → prevent runaway complement
Loss-of-function mutations (allow unopposed complement activation):
- CFH (most common) — key plasma downregulator; heterozygotes — abnormal CFH inactivates normal CFH
- CFI — cleaves C3b and C4b
- MCP / CD46 — cell-surface cofactor for CFI (best prognosis; high recurrence post-transplant)
- Thrombomodulin
Gain-of-function mutations (increase convertase activity):
- C3 — more C3b generated
- Factor B (CFB) — more stable C3bBb convertase
Death/ESRD >50% + recurrence >75%: CFH, CFI, CFB, thrombomodulin mutations
Best prognosis: MCP (CD46) mutations
Autoimmune aHUS: anti-CFH antibodies (~6% of aHUS; mostly <16 years)
Triggers in carriers: Pregnancy (postpartum — 20% of aHUS), OCP (CFH: 8%; CFI: 20%), infections
Eculizumab NON-responsive: DGKE, MMACHC, INF2 mutations; C5 polymorphism p.R885H
=== RENAL HISTOPATHOLOGY ===
Light Microscopy:
- Microthrombi in glomerular capillaries, afferent arterioles, interlobular arteries
- Endothelial swelling and detachment from GBM
- Widening of subendothelial space (fluffy material accumulation)
- GBM duplication — "double contour" / tram-track (new BM forms over injured subendothelial zone)
- Mesangiolysis — lysis of mesangial cells + loose mesangial matrix
- Onion-skin arteriolar lesion — concentric myointimal hyperplasia (more in aHUS/chronic)
- Patchy cortical necrosis — in severe cases
- Crescent formation — uncommon
- FSGS — late sequela (chronic hypertension + progressive CKD)
Pattern:
- STEC-HUS / young children: glomerular injury predominates
- Idiopathic/familial/adult aHUS: arteriolar injury predominates → secondary glomerular ischemia
Electron Microscopy:
- Endothelial swelling and detachment from basement membrane
- Subendothelial fluffy electron-lucent material (widened subendothelial space)
- Intraluminal platelet thrombi
- Similar EM appearance to: scleroderma, malignant nephrosclerosis, chronic transplant rejection, calcineurin inhibitor nephrotoxicity
Immunofluorescence:
- NEGATIVE for immunoglobulins (IgG, IgA, IgM) — KEY
- Variable fibrinogen in glomeruli and arterioles
- Some C3 staining may be present (especially aHUS)
- No immune complex deposition
- Cannot differentiate TMA causes by pathology alone — clinical/lab context is essential
=== PERIPHERAL BLOOD SMEAR ===
- Schistocytes (fragmented RBCs) = hallmark of MAHA
- Forms: helmet cells, burr cells, triangle cells
- Mechanism: microvascular thrombi → shear stress → mechanical RBC fragmentation → intravascular hemolysis
- Schistocyte count: MORE in TTP (>3/HPF) than in HUS
=== LABORATORY PATHOLOGY ===
- LDH: markedly elevated (hemolysis + tissue injury)
- Haptoglobin: decreased/absent (intravascular hemolysis)
- Indirect bilirubin: mildly elevated
- Reticulocyte count: elevated
- Direct Coombs test: NEGATIVE (non-immune, mechanical hemolysis)
- Platelets: <100,000/uL
- PT/aPTT: NORMAL — key, differentiates from DIC
- Fibrinogen: Normal — key, differentiates from DIC
- ADAMTS13: NORMAL — key, differentiates from TTP (TTP = <10%)
- Creatinine: elevated, subacutely worsening
- Urinalysis: hematuria, proteinuria, granular casts
- C3: low in ~50% STEC-HUS; persistent low C3 = poor prognosis
=== HUS vs TTP vs DIC ===
Feature | HUS | TTP | DIC
Dominant organ | Kidney (AKI) | Brain (neuro) | Multi-organ
Fever | Absent | Present | Variable
Schistocytes | Present | Present (>3/HPF)| Present
ADAMTS13 | Normal | <10% (deficient)| Normal
PT/aPTT | Normal | Normal | Prolonged
Fibrinogen | Normal | Normal | Decreased
D-dimers | Normal/mild up | Normal | Elevated
Treatment | Supportive/eculi | Plasmapheresis | Treat cause
=== MICROTHROMBI DISTRIBUTION ===
HUS: primarily kidneys (glomerular capillaries + afferent arterioles)
TTP: kidneys, pancreas, heart, adrenals, brain (more widespread)
Neurological involvement in HUS: <50% of cases
=== CAUSES OF MAHA (MCQ LIST) ===
HUS, TTP, DIC, malignant hypertension, SLE, preeclampsia/HELLP, disseminated cancer, prosthetic cardiac valves, scleroderma renal crisis, APS, drugs (mitomycin C, gemcitabine)
=== COMPLEMENT CASCADE — HIGH-YIELD ===
Alternative pathway C3 convertase = C3bBb (C3b + Factor B, cleaved by Factor D)
AP amplification loop = ~80% of all complement activity
Terminal pathway: C5 convertase → C5a (anaphylatoxin) + C5b → C5b-9 (MAC)
Eculizumab: binds C5 → blocks C5a + C5b → prevents MAC → stops endothelial/platelet damage
Ravulizumab: same mechanism, extended half-life, FDA approved for aHUS
=== DRUGS CAUSING SECONDARY HUS ===
- Mitomycin C: most common chemotherapy cause; dose-dependent; onset 4-8 weeks after last dose
- Gemcitabine: NOT dose-dependent; gastric/lung/colorectal/pancreatic/breast cancers
- Cyclosporine/Tacrolimus: post-transplant TMA
- VEGF inhibitors (bevacizumab): endothelial injury
- Valacyclovir (high dose in HIV): rare but fatal
- OCP: trigger in complement mutation carriers
=== PNEUMOCOCCAL HUS ===
- S. pneumoniae → neuraminidase cleaves sialic acid → exposes Thomsen-Friedenreich (T) antigen on RBCs, platelets, endothelium
- Natural IgM anti-T antibodies bind → hemolysis + endothelial injury
- AVOID FFP/plasma exchange — FFP contains anti-T antibodies → worsens disease
- Treatment: antibiotics + supportive care only
=== PROGNOSIS — PATHOLOGICAL CORRELATES ===
- CFH, CFI, CFB, thrombomodulin mutations: Death/ESRD >50%; recurrence >75%
- MCP (CD46) mutations: best prognosis; high recurrence post-transplant
- Anti-CFH antibodies: good response to immunosuppression
- Cortical necrosis on biopsy: severe; may not recover
- FSGS as late sequela: progressive CKD
- STEC-HUS with dialysis: most recover in weeks; long-term (15-25 yrs) not uniformly favorable
- Persistent low C3 in STEC-HUS: poor prognosis
=== TREATMENT — KEY POINTS ===
- STEC-HUS: supportive only — NO antibiotics (accelerate Stx release), plasma exchange NOT effective
- aHUS: Eculizumab (first-line, FDA-approved) — blocks C5; mandatory N. meningitidis vaccination before starting
- TTP: Plasma exchange (life-saving) — removes anti-ADAMTS13 antibodies + replaces ADAMTS13
- Rituximab: refractory TTP, autoimmune aHUS, chemotherapy-induced HUS
- Caplacizumab: TTP — anti-vWF nanobody, blocks vWF-platelet interaction
- Pneumococcal HUS: NO FFP
=== MUST-KNOW MCQ ANCHORS ===
- Most common cause of AKI requiring dialysis in children 1-5 years = HUS
- Shiga toxin receptor = Gb3 (globotriaosylceramide)
- More nephrotoxic Stx = Stx2
- Stx A subunit = inhibits ribosome; B subunit = binds Gb3
- Most common STEC serotype (Europe/N. America) = E. coli O157:H7
- Largest HUS outbreak (2011, Germany) = E. coli O104:H4 (fenugreek sprouts)
- Shiga HUS in Asia/Africa = Shigella dysenteriae type 1
- Coombs test in HUS = NEGATIVE
- IF on renal biopsy = negative for Ig (variable fibrinogen; some C3)
- EM hallmark = subendothelial fluffy material + endothelial swelling + intraluminal thrombi
- Cannot differentiate TMA causes on biopsy alone
- Most important complement regulator in aHUS = Factor H (CFH)
- Eculizumab target = C5
- Mandatory pre-eculizumab vaccination = N. meningitidis
- Avoid antibiotics in STEC-HUS = accelerate Stx release
- Avoid FFP in pneumococcal HUS = contains anti-T antibodies
- Most common chemotherapy causing HUS = Mitomycin C
- % STEC progressing to HUS = 5-10%
- OCP trigger: CFH = 8%, CFI = 20%
- Pregnancy-associated aHUS = postpartum (20% of all aHUS)
- Eculizumab non-responsive = DGKE, MMACHC, INF2, C5 p.R885H
Still showing reverse. Front should show HUS heading. The back card should show the rest of the summary.
<br> tags instead, with #html:true.#separator:tab
#html:true
#notetype:Basic
Hemolytic Uremic Syndrome — Pathology (NEET PG 2026) <b>=== DEFINITION / TRIAD ===</b><br>HUS = Thrombotic Microangiopathy (TMA) with:<br>1. Microangiopathic Hemolytic Anemia (MAHA)<br>2. Thrombocytopenia<br>3. Acute Kidney Injury (AKI) — dominant organ in HUS<br><br><b>=== CLASSIFICATION ===</b><br>Typical HUS (D+ HUS / STEC-HUS):<br>- Cause: Shiga toxin-producing E. coli (O157:H7 — most common in Europe/N. America) or Shigella dysenteriae type 1 (Asia/Africa)<br>- Age: Children <5 years; bloody diarrhea prodrome (~60%)<br>- >90% of childhood HUS<br><br>Atypical HUS (D- HUS / aHUS):<br>- Cause: Genetic or autoimmune dysregulation of alternative complement pathway<br>- Any age; ~67% in childhood; NO diarrhea prodrome<br><br>Secondary HUS: Drugs, malignancy, pregnancy, HIV, transplant<br><br><b>=== PATHOGENESIS — TYPICAL HUS ===</b><br>1. STEC colonizes gut via intimin (eaeA gene) — attaching/effacing lesion<br>2. Shiga toxin (Stx) produced — Stx2 more nephrotoxic than Stx1<br>3. Toxin enters circulation attached to leukocytes, RBCs, platelets<br>4. B subunit (pentameric) binds Gb3 (globotriaosylceramide) on renal endothelium → internalized via clathrin-coated pits → retrograde transport via Golgi → ER<br>5. A subunit inhibits ribosomal protein synthesis → endothelial cell death<br>6. Endothelial injury → platelet activation → microvascular thrombosis<br><br>Why kidney most affected: Glomerular endothelium has highest Gb3 density (also podocytes, mesangial cells, tubular epithelium)<br>Why children more affected: Higher Gb3 expression; adults have anti-Stx antibodies<br>Low-dose Stx: activates endothelium → increased endothelin, decreased NO → vasoconstriction + platelet adhesion<br>High-dose Stx: direct endothelial cell death<br>Stx releases IL-1, TNF-alpha, IL-6 → procoagulant milieu<br>Complement (AP) activated in ~50% STEC-HUS (low C3); persistent low C3 = poor prognosis<br><br><b>=== PATHOGENESIS — ATYPICAL HUS ===</b><br>Core defect: Uncontrolled alternative complement pathway (C3 convertase = C3bBb)<br><br>Normal regulators (lost in aHUS):<br>- CFH (most common) — degrades C3b; heterozygotes: abnormal CFH inactivates normal CFH<br>- CFI — cleaves C3b and C4b<br>- MCP / CD46 — cell-surface cofactor for CFI (best prognosis; high recurrence post-transplant)<br>- Thrombomodulin<br><br>Gain-of-function mutations:<br>- C3 — more C3b generated<br>- Factor B (CFB) — more stable C3bBb convertase<br><br>Death/ESRD >50% + recurrence >75%: CFH, CFI, CFB, thrombomodulin mutations<br>Best prognosis: MCP (CD46) mutations<br>Autoimmune aHUS: anti-CFH antibodies (~6%; mostly <16 years)<br>Triggers: Pregnancy postpartum (20% of aHUS), OCP (CFH: 8%; CFI: 20%), infections<br>Eculizumab NON-responsive: DGKE, MMACHC, INF2 mutations; C5 p.R885H<br><br><b>=== RENAL HISTOPATHOLOGY ===</b><br><b>Light Microscopy:</b><br>- Microthrombi in glomerular capillaries, afferent arterioles, interlobular arteries<br>- Endothelial swelling and detachment from GBM<br>- Widening of subendothelial space (fluffy material)<br>- GBM duplication — double contour/tram-track (new BM over injured zone)<br>- Mesangiolysis — lysis of mesangial cells + loose mesangial matrix<br>- Onion-skin arteriolar lesion — concentric myointimal hyperplasia (aHUS/chronic)<br>- Patchy cortical necrosis — severe cases<br>- Crescent formation — uncommon<br>- FSGS — late sequela (chronic hypertension + progressive CKD)<br><br>Pattern: STEC-HUS/children = glomerular injury predominates; aHUS/adult = arteriolar injury predominates → secondary glomerular ischemia<br><br><b>Electron Microscopy:</b><br>- Endothelial swelling + detachment from BM<br>- Subendothelial fluffy electron-lucent material<br>- Intraluminal platelet thrombi<br>- Similar to: scleroderma, malignant nephrosclerosis, chronic transplant rejection, calcineurin inhibitor nephrotoxicity<br><br><b>Immunofluorescence:</b><br>- NEGATIVE for immunoglobulins (IgG, IgA, IgM) — KEY<br>- Variable fibrinogen; some C3 (especially aHUS)<br>- No immune complex deposition<br>- Cannot differentiate TMA causes on pathology alone<br><br><b>=== PERIPHERAL BLOOD SMEAR ===</b><br>Schistocytes = hallmark of MAHA (also: helmet cells, burr cells, triangle cells)<br>Mechanism: microvascular thrombi → shear stress → mechanical RBC fragmentation → intravascular hemolysis<br>Schistocyte count: MORE in TTP (>3/HPF) than HUS<br><br><b>=== LABORATORY PATHOLOGY ===</b><br>- LDH: markedly elevated<br>- Haptoglobin: decreased/absent<br>- Indirect bilirubin: mildly elevated<br>- Reticulocyte count: elevated<br>- Direct Coombs test: NEGATIVE (non-immune, mechanical hemolysis)<br>- Platelets: <100,000/uL<br>- PT/aPTT: NORMAL (differentiates from DIC)<br>- Fibrinogen: Normal (differentiates from DIC)<br>- ADAMTS13: NORMAL (differentiates from TTP where <10%)<br>- Urinalysis: hematuria, proteinuria, granular casts<br>- C3: low in ~50% STEC-HUS; persistent low = poor prognosis<br><br><b>=== HUS vs TTP vs DIC ===</b><br>Dominant organ: HUS = Kidney | TTP = Brain | DIC = Multi-organ<br>Fever: HUS = Absent | TTP = Present | DIC = Variable<br>PT/aPTT: HUS = Normal | TTP = Normal | DIC = Prolonged<br>Fibrinogen: HUS = Normal | TTP = Normal | DIC = Decreased<br>D-dimers: HUS = Normal | TTP = Normal | DIC = Elevated<br>ADAMTS13: HUS = Normal | TTP = <10% | DIC = Normal<br>Treatment: HUS = Supportive/eculizumab | TTP = Plasmapheresis | DIC = Treat cause<br><br><b>=== MICROTHROMBI DISTRIBUTION ===</b><br>HUS: kidneys (glomerular capillaries + afferent arterioles)<br>TTP: kidneys, pancreas, heart, adrenals, brain (more widespread)<br>Neurological involvement in HUS: <50%<br><br><b>=== COMPLEMENT CASCADE ===</b><br>AP C3 convertase = C3bBb (C3b + Factor B, cleaved by Factor D)<br>AP amplification = ~80% of all complement activity<br>Terminal: C5 convertase → C5a + C5b → MAC (C5b-9)<br>Eculizumab: binds C5 → blocks C5a + C5b → prevents MAC<br>Ravulizumab: same mechanism, extended half-life, FDA approved for aHUS<br><br><b>=== DRUGS CAUSING SECONDARY HUS ===</b><br>- Mitomycin C: most common; dose-dependent; onset 4-8 weeks after last dose<br>- Gemcitabine: NOT dose-dependent; gastric/lung/colorectal/pancreatic/breast<br>- Cyclosporine/Tacrolimus: post-transplant TMA<br>- VEGF inhibitors (bevacizumab): endothelial injury<br>- Valacyclovir high dose (HIV): rare but fatal<br>- OCP: trigger in complement mutation carriers<br><br><b>=== PNEUMOCOCCAL HUS ===</b><br>S. pneumoniae → neuraminidase cleaves sialic acid → exposes Thomsen-Friedenreich (T) antigen<br>Natural IgM anti-T antibodies → hemolysis + endothelial injury<br>AVOID FFP/plasma exchange (FFP contains anti-T antibodies → worsens)<br>Treatment: antibiotics + supportive only<br><br><b>=== PROGNOSIS ===</b><br>- CFH, CFI, CFB, thrombomodulin: Death/ESRD >50%; recurrence >75%<br>- MCP (CD46): best prognosis; high recurrence post-transplant<br>- Cortical necrosis on biopsy: may not recover<br>- FSGS late sequela: progressive CKD<br>- STEC-HUS: most recover in weeks with dialysis<br><br><b>=== TREATMENT KEY POINTS ===</b><br>- STEC-HUS: supportive only; NO antibiotics (accelerate Stx release); plasma exchange NOT effective<br>- aHUS: Eculizumab (first-line, FDA-approved, blocks C5); mandatory N. meningitidis vaccination before<br>- TTP: Plasma exchange (life-saving)<br>- Rituximab: refractory TTP, autoimmune aHUS, chemo-induced HUS<br>- Caplacizumab: TTP (anti-vWF nanobody)<br>- Pneumococcal HUS: NO FFP<br><br><b>=== MCQ ANCHORS ===</b><br>- Most common cause of AKI/dialysis in children 1-5 yrs = HUS<br>- Shiga toxin receptor = Gb3 (globotriaosylceramide)<br>- More nephrotoxic = Stx2<br>- A subunit = inhibits ribosome; B subunit = binds Gb3<br>- Most common STEC (Europe/N. America) = E. coli O157:H7<br>- Largest outbreak (2011 Germany) = E. coli O104:H4 (fenugreek sprouts)<br>- Asia/Africa = Shigella dysenteriae type 1<br>- Coombs test = NEGATIVE<br>- IF on biopsy = negative for Ig (variable fibrinogen; some C3)<br>- EM hallmark = subendothelial fluffy material + endothelial swelling + intraluminal thrombi<br>- Pathology alone cannot differentiate TMA causes<br>- Most important complement regulator = Factor H (CFH)<br>- Eculizumab target = C5<br>- Pre-eculizumab vaccination = N. meningitidis<br>- Avoid antibiotics in STEC-HUS = accelerate Stx release<br>- Avoid FFP in pneumococcal HUS = anti-T antibodies in FFP<br>- Most common chemo causing HUS = Mitomycin C<br>- % STEC progressing to HUS = 5-10%<br>- OCP trigger: CFH = 8%, CFI = 20%<br>- Pregnancy aHUS = postpartum (20% of all aHUS)<br>- Eculizumab non-responsive = DGKE, MMACHC, INF2, C5 p.R885H
Hemolytic Uremic Syndrome — Pathology (NEET PG 2026)) is separated from the back by a single tab character, and the entire back uses <br> tags instead of real line breaks. This is the only reliable way to guarantee AnkiDroid reads it as one card with front on the left of the tab and back on the right — because real line breaks in the file confuse the importer into thinking a new card or field has started.I also want you to make 20 very high-yield Anki cards for the topic Pathology of Hemolytic Uremic Syndrome, covering almost 100% of pathology questions that can be asked in NEET PG 2026 MCQs. These cards should help me to answer MCQ questions. It should be in text format, so that I can copy and paste to Ankita text app and then import to AnkiDroid app. The front part should be the question and the back part should be the answer.
#separator:tab
#html:true
#notetype:Basic
What is the classic triad of Hemolytic Uremic Syndrome (HUS)? <b>Triad of HUS:</b><br>1. Microangiopathic Hemolytic Anemia (MAHA)<br>2. Thrombocytopenia<br>3. Acute Kidney Injury (AKI)<br><br><b>Key:</b> AKI is the dominant organ involvement in HUS (unlike TTP where brain dominates)
What is the most common cause of HUS in children and what organism is responsible? <b>Most common cause:</b> Shiga toxin-producing E. coli (STEC)<br><b>Most common serotype:</b> E. coli O157:H7 (Europe/North America)<br><b>Also caused by:</b> Shigella dysenteriae type 1 (Asia/Africa)<br><br><b>Key facts:</b><br>- >90% of childhood HUS is STEC-HUS<br>- Most common cause of AKI requiring dialysis in children aged 1-5 years<br>- Prodrome: bloody diarrhea (~60% of cases), 3 days post-exposure; HUS develops ~7 days later<br>- 5-10% of STEC infections progress to HUS
What is the receptor for Shiga toxin on renal endothelium and why is the kidney most affected? <b>Receptor:</b> Gb3 (Globotriaosylceramide) — a glycolipid receptor<br><br><b>Why kidney is most affected:</b><br>- Glomerular endothelium expresses the HIGHEST density of Gb3<br>- Gb3 also on podocytes, mesangial cells, and tubular epithelial cells<br><br><b>Why children more affected than adults:</b><br>- Higher Gb3 expression on renal endothelium in children<br>- Adults have protective circulating anti-Stx antibodies
What are the A and B subunits of Shiga toxin and what do they do? <b>B subunit (pentameric):</b><br>- Binds to Gb3 receptor on cell surface<br>- Mediates internalization via clathrin-coated pits<br>- Retrograde transport: Golgi → Endoplasmic Reticulum<br><br><b>A subunit:</b><br>- Inhibits ribosomal protein synthesis (cleaves 28S rRNA)<br>- Causes endothelial cell death<br><br><b>Which Stx is more nephrotoxic?</b> Stx2 > Stx1
What are the light microscopy findings on renal biopsy in HUS? <b>Light Microscopy — TMA in HUS:</b><br>- Microthrombi in glomerular capillaries, afferent arterioles, interlobular arteries<br>- Endothelial swelling and detachment from GBM<br>- Widening of subendothelial space (fluffy material)<br>- GBM duplication — "double contour" / tram-track appearance<br>- Mesangiolysis — lysis of mesangial cells + loose mesangial matrix<br>- Onion-skin arteriolar lesion — concentric myointimal hyperplasia (aHUS/chronic)<br>- Patchy cortical necrosis — severe cases<br>- FSGS — late sequela (chronic hypertension + CKD)<br><br><b>Pattern:</b> STEC-HUS/children = glomerular injury predominates; aHUS/adults = arteriolar injury predominates
What are the electron microscopy (EM) findings in HUS renal biopsy? <b>EM findings in TMA/HUS:</b><br>- Endothelial cell swelling and detachment from basement membrane<br>- Accumulation of fluffy electron-lucent material in subendothelial space (widened subendothelial space)<br>- Intraluminal platelet thrombi<br>- Partial or complete obstruction of vessel lumina<br><br><b>Similar EM appearance to:</b><br>- Scleroderma renal crisis<br>- Malignant nephrosclerosis<br>- Chronic transplant rejection<br>- Calcineurin inhibitor (cyclosporine/tacrolimus) nephrotoxicity
What are the immunofluorescence (IF) findings on renal biopsy in HUS? <b>Immunofluorescence in HUS/TMA:</b><br>- NEGATIVE for immunoglobulins (IgG, IgA, IgM) — KEY finding<br>- Variable fibrinogen deposits in glomeruli and arterioles<br>- Some C3 staining may be present (especially in aHUS)<br>- NO immune complex deposition<br><br><b>Key MCQ point:</b> IF is negative for Ig — this differentiates TMA from immune complex GN (MPGN, IgAN, lupus nephritis)<br><br><b>Bonus:</b> Pathology alone CANNOT differentiate the various causes of TMA — clinical and lab context is essential
What are the peripheral blood smear findings in HUS and what is the mechanism of hemolysis? <b>Peripheral smear findings:</b><br>- Schistocytes (fragmented RBCs) = hallmark of MAHA<br>- Morphological variants: helmet cells, burr cells, triangle cells<br><br><b>Mechanism of hemolysis:</b><br>Microvascular thrombi → luminal narrowing → shear stress on passing RBCs → mechanical fragmentation → intravascular hemolysis → schistocytes<br><br><b>Schistocyte count comparison:</b><br>- TTP: MORE schistocytes (>3/high power field)<br>- HUS: fewer schistocytes than TTP<br><br><b>Direct Coombs test:</b> NEGATIVE (non-immune, mechanical hemolysis)
What laboratory findings help differentiate HUS from TTP and DIC? <b>Key differentiating labs:</b><br><br>Feature | HUS | TTP | DIC<br>ADAMTS13 | Normal | <10% (deficient) | Normal<br>PT/aPTT | NORMAL | Normal | PROLONGED<br>Fibrinogen | Normal | Normal | DECREASED<br>D-dimers | Normal/mild up | Normal | ELEVATED<br>Coombs test | Negative | Negative | Negative<br>Dominant organ | Kidney | Brain | Multi-organ<br>Fever | ABSENT | Present | Variable<br><br><b>Key rule:</b> Normal PT/aPTT + normal fibrinogen = NOT DIC; Normal ADAMTS13 = NOT TTP
What is the most common genetic mutation in atypical HUS (aHUS) and what is the mechanism? <b>Most common mutation:</b> Complement Factor H (CFH) — loss of function<br><br><b>Mechanism:</b><br>- CFH is the main plasma downregulator of the alternative complement pathway<br>- CFH normally degrades C3b, preventing runaway C3 convertase (C3bBb) activity<br>- In heterozygotes: abnormal CFH complexes with normal CFH → inactivates it<br>- Result: Unopposed C3b activity → uncontrolled complement activation → endothelial injury → TMA<br><br><b>Other loss-of-function mutations in aHUS:</b><br>- CFI (cleaves C3b and C4b)<br>- MCP/CD46 (cell-surface cofactor for CFI) — best prognosis<br>- Thrombomodulin<br><br><b>Gain-of-function mutations:</b> C3, Factor B (CFB) — more stable C3 convertase
What is the prognosis of different complement mutations in aHUS? <b>Poor prognosis (Death/ESRD >50%; recurrence >75%):</b><br>- CFH mutations<br>- CFI mutations<br>- CFB mutations (gain of function)<br>- Thrombomodulin mutations<br><br><b>Best prognosis:</b><br>- MCP (CD46) mutations — good renal survival but HIGH recurrence rate post-transplant<br><br><b>Autoimmune aHUS (anti-CFH antibodies):</b><br>- ~6% of aHUS cases<br>- Mostly diagnosed <16 years<br>- Good response to immunosuppression<br><br><b>Triggers in carriers:</b> Pregnancy postpartum (20% of aHUS), OCP (CFH: 8%; CFI: 20%), infections
What is the mechanism of Pneumococcal HUS and why is FFP contraindicated? <b>Organism:</b> Streptococcus pneumoniae<br><br><b>Mechanism:</b><br>1. S. pneumoniae produces neuraminidase<br>2. Neuraminidase cleaves sialic acid from RBCs, platelets, and glomerular endothelium<br>3. This exposes the Thomsen-Friedenreich (T) antigen (normally hidden)<br>4. Naturally occurring IgM anti-T antibodies (present in all humans) bind to exposed T antigen<br>5. Result: hemolysis + endothelial injury → HUS<br><br><b>Why FFP is CONTRAINDICATED:</b><br>FFP contains IgM anti-T antibodies → binding to exposed T antigen → WORSENS hemolysis and endothelial injury<br><br><b>Treatment:</b> Antibiotics + supportive care only (NO plasma exchange, NO FFP)
What is the mechanism of action of Eculizumab in aHUS and what vaccination is mandatory before starting it? <b>Eculizumab mechanism:</b><br>- Recombinant humanized monoclonal antibody<br>- Binds to complement protein C5<br>- Blocks cleavage of C5 into C5a (anaphylatoxin) and C5b<br>- Prevents formation of Membrane Attack Complex (MAC = C5b-9)<br>- Stops endothelial damage and platelet activation<br><br><b>Ravulizumab:</b> Same mechanism, extended half-life, also FDA-approved for aHUS<br><br><b>Mandatory vaccination BEFORE starting:</b> Neisseria meningitidis<br>(Reason: terminal complement pathway is blocked → cannot lyse encapsulated organisms → high risk of meningococcal sepsis)<br><br><b>Efficacy:</b> ~85% of aHUS patients become disease-free
Why are antibiotics CONTRAINDICATED in STEC-HUS? <b>Answer:</b> Antibiotics are contraindicated in STEC-HUS because they cause lysis of E. coli bacteria → accelerated release of large amounts of Shiga toxin into the circulation → worsening of HUS<br><br><b>Treatment of STEC-HUS is:</b> Supportive only<br>- Fluid resuscitation<br>- Blood pressure control<br>- Dialysis if needed<br><br><b>Plasma exchange:</b> NOT effective in STEC-HUS (unlike TTP where it is life-saving)<br><br><b>Eculizumab in STEC-HUS:</b> Controversial; not standard of care
What is the distribution of microthrombi in HUS vs TTP on pathology? <b>HUS:</b><br>- Microthrombi predominantly in KIDNEYS<br>- Glomerular capillaries + afferent arterioles + interlobular arteries<br>- Neurological involvement in <50% of cases<br><br><b>TTP:</b><br>- Microthrombi more WIDESPREAD<br>- Predominantly: kidneys, pancreas, heart, adrenals, brain<br>- Neurological involvement is the dominant feature<br><br><b>Key MCQ rule:</b><br>AKI dominant → think HUS<br>Neurological signs dominant → think TTP<br><br><b>Fever:</b> Absent in HUS; Present in TTP (part of classic pentad)
What drugs most commonly cause secondary HUS/TMA? <b>Most common chemotherapy cause:</b> Mitomycin C (dose-dependent; onset 4-8 weeks after last dose)<br><br><b>Other chemotherapy drugs:</b><br>- Gemcitabine (NOT dose-dependent; cancers: gastric, lung, colorectal, pancreatic, breast)<br>- Cisplatin, bleomycin (rare)<br>- VEGF inhibitors (bevacizumab) — endothelial injury<br><br><b>Immunosuppressants:</b><br>- Cyclosporine / Tacrolimus (calcineurin inhibitors) — post-transplant TMA<br><br><b>Others:</b><br>- Quinine (immune-mediated)<br>- Valacyclovir high dose (HIV patients) — rare but fatal<br>- Oral contraceptives — trigger in complement mutation carriers<br>- Cocaine<br><br><b>Treatment of chemo-induced HUS:</b> Rituximab (effective); plasma exchange generally poor outcome
What are the causes of Microangiopathic Hemolytic Anemia (MAHA) — the complete MCQ list? <b>Causes of MAHA (schistocytes on smear):</b><br><br><b>TMA-related:</b><br>- HUS (typical and atypical)<br>- TTP<br>- DIC<br><br><b>Vascular/pressure:</b><br>- Malignant hypertension<br>- Scleroderma renal crisis<br>- Preeclampsia / HELLP syndrome<br><br><b>Immune/autoimmune:</b><br>- SLE (lupus)<br>- Antiphospholipid antibody syndrome (APS)<br><br><b>Malignancy:</b><br>- Disseminated cancer (gastric, lung, breast)<br><br><b>Mechanical:</b><br>- Prosthetic cardiac valves (turbulent flow)<br><br><b>Drugs:</b><br>- Mitomycin C, gemcitabine, cyclosporine, quinine<br><br><b>Infections:</b><br>- HIV, bone marrow transplant-associated TMA
What is the E. coli serotype responsible for the largest recorded HUS outbreak and where did it occur? <b>Largest recorded HUS outbreak:</b><br>- Year: May 2011<br>- Location: Germany<br>- Organism: E. coli O104:H4<br>- Source: Fenugreek sprouts contaminated with fecal material (seeds from Egypt)<br>- Notable: O104:H4 was NOT found in cattle feces from the outbreak area — unusual STEC strain<br>- HUS progression rate: ~20% (much higher than usual 5-10%)<br><br><b>Usual most common STEC serotype (Europe/North America):</b> E. coli O157:H7<br><b>Asia/Africa cause:</b> Shigella dysenteriae type 1
What is the role of ADAMTS13 in TTP vs HUS and how does Shiga toxin interact with it? <b>ADAMTS13 (A disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13):</b><br>- Normal function: Cleaves ultra-large von Willebrand factor (vWF) multimers into smaller, less adhesive sizes<br><br><b>In TTP:</b><br>- ADAMTS13 activity <10% (severe deficiency)<br>- Acquired: autoantibodies against ADAMTS13 (most common)<br>- Inherited: genetic mutations in ADAMTS13 gene (rare)<br>- Result: Ultra-large vWF multimers → spontaneous platelet aggregation → widespread microvascular thrombosis<br><br><b>In HUS:</b><br>- ADAMTS13 level is NORMAL (>10%)<br>- KEY differentiating point from TTP<br><br><b>Shiga toxin interaction:</b><br>- Stx inhibits endothelial production of ADAMTS13<br>- This contributes to vWF accumulation and platelet aggregation in STEC-HUS
What are the eculizumab-non-responsive forms of aHUS and what are the mutations involved? <b>Eculizumab NON-responsive aHUS — mutations:</b><br><br>1. <b>DGKE</b> (Diacylglycerol kinase epsilon) — non-complement pathway; affects coagulation<br>2. <b>MMACHC</b> (Methylmalonic aciduria and homocystinuria type C) — metabolic; cobalamin C defect<br>3. <b>INF2</b> (Inverted formin 2) — cytoskeletal protein<br>4. <b>C5 polymorphism p.R885H</b> — eculizumab binding site on C5 is altered → drug cannot bind<br><br><b>Key concept:</b> These patients have aHUS-like TMA but through non-complement mechanisms, so blocking C5 does not help<br><br><b>Contrast with eculizumab-responsive:</b> CFH, CFI, CFB, C3, MCP, thrombomodulin mutations, anti-CFH antibodies (~85% respond)
- Copy everything above from
#separator:tabto the last line - Paste into a plain text editor (Notepad, Google Keep, etc.) and save as
HUS_MCQ_Cards.txt - In AnkiDroid → Menu → Import → select the
.txtfile - Confirm: Separator = Tab, HTML = enabled, Notetype = Basic
- Import — 20 cards will be created, each with a question on the front and answer on the back