Rassmussens Encephalitis
Reading File
search_medical_text
Rasmussen's encephalitis clinical features diagnosis pathophysiology
search_medical_text
Rasmussen encephalitis treatment management hemispherectomy immunotherapy
search_medical_image
Rasmussen encephalitis MRI brain imaging
GradeResponse
GradeResponse
GradeResponse
The database retrieved limited text, so I'll supplement with comprehensive knowledge.
Rasmussen's Encephalitis (Rasmussen Syndrome)
Rasmussen's encephalitis (RE) is a rare, chronic, progressive inflammatory brain disease predominantly affecting one cerebral hemisphere. It is classically associated with intractable focal epilepsy, progressive neurological deficits, and unihemispheric atrophy.
Epidemiology
- Incidence: ~2.4 per 10 million per year
- Onset typically in childhood (median age ~6–8 years), though adult-onset cases (~10%) are recognized
- No sex predilection
- No geographic or ethnic clustering
Pathophysiology
The etiology remains incompletely understood but is considered autoimmune in nature, with T-cell-mediated cytotoxic destruction of neurons and astrocytes:
- CD8+ cytotoxic T lymphocytes are the primary effectors, found in direct apposition to neurons and astrocytes in affected cortex
- Microglial nodules and perivascular lymphocytic cuffing are hallmark histological findings
- Anti-GluR3 (GluA3) antibodies were historically implicated, but their pathogenic role is debated; anti-GluR3 is not consistently present
- Anti-NMDA receptor antibodies and other neuronal antibodies have been found in subsets
- A viral trigger (CMV, EBV, enterovirus) has been hypothesized but never proven
- The process is self-limited to one hemisphere, which remains unexplained
Clinical Stages (Bien et al. 2002)
RE progresses through three recognized stages:
| Stage | Name | Features |
|---|---|---|
| 1 | Prodromal | Low seizure frequency, mild or no deficits; may last months to years |
| 2 | Acute | Frequent focal seizures, epilepsia partialis continua (EPC), hemiparesis, cognitive decline |
| 3 | Residual | Permanent hemiplegia, fixed neurological deficits, reduced seizure frequency |
Clinical Features
Seizures
- Focal motor seizures are the hallmark, often simple partial (conscious patient)
- Epilepsia Partialis Continua (EPC): continuous focal clonic jerking of one body part for hours to days — a near-pathognomonic feature (~50–70% of patients)
- Seizures are typically medically refractory
- Secondary generalization may occur
Neurological Deficits (progressive)
- Contralateral hemiparesis (most common)
- Hemianopia (contralateral visual field loss)
- Aphasia (if dominant hemisphere affected)
- Cognitive decline / dementia
- Hemisensory deficits
Diagnosis
Diagnosis is clinical + radiological + histopathological. The European Consensus Statement (2005) provides diagnostic criteria with two pathways:
Part A (Clinical + MRI — no biopsy needed if all present)
- Focal seizures (with or without EPC) and unilateral cortical deficit
- EEG with unihemispheric slowing or epileptic activity, contralateral to the clinical deficit
- MRI showing unihemispheric focal cortical atrophy with T2/FLAIR hyperintensity in gray or white matter, or T2 signal in ipsilateral caudate head
Part B (Less typical presentations — requires biopsy confirmation)
- Focal seizures or EPC with or without unilateral cortical deficit
- MRI showing progressive unihemispheric focal cortical atrophy
- Histopathology: T-cell-dominated encephalitis with activated microglial cells ± reactive astrogliosis; multiple macrophages, perivascular lymphocytes
MRI Findings
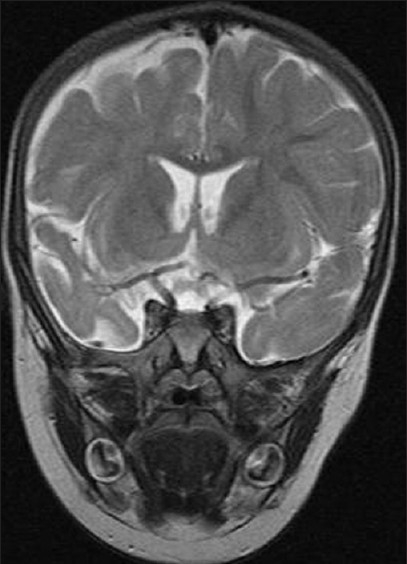
T2-weighted coronal MRI showing characteristic unihemispheric cortical atrophy in Rasmussen's encephalitis: cortical thinning, sulcal widening, T2 hyperintensity in cortex and subcortical white matter, and ex-vacuo dilatation of the ipsilateral lateral ventricle.
Sequential MRI findings (evolving over months–years):
- Early: focal cortical swelling or T2 hyperintensity, ipsilateral caudate T2 signal
- Progressive: unihemispheric cortical atrophy, white matter atrophy
- Late: severe ipsilateral hemispheric atrophy with compensatory contralateral hypertrophy
- FDG-PET: unihemispheric hypometabolism (useful in early/atypical cases)
Differential Diagnosis
| Condition | Distinguishing Features |
|---|---|
| Focal cortical dysplasia | Static lesion on MRI; no progressive atrophy |
| FIRES / febrile infection-related epilepsy | Bilateral involvement, acute onset post-fever |
| Anti-NMDAR encephalitis | Bilateral, psychiatric prodrome, specific antibody |
| Hemimegalencephaly | Enlarged (not atrophic) hemisphere |
| Sturge-Weber syndrome | Port-wine stain, leptomeningeal angioma |
| MELAS | Stroke-like episodes, mitochondrial pattern |
| Lafora disease | Progressive myoclonic epilepsy, bilateral |
Treatment
RE is divided into two treatment domains: disease-modifying immunotherapy and surgical intervention.
Immunotherapy (Slows Progression, Rarely Curative)
| Agent | Mechanism / Route | Evidence Level |
|---|---|---|
| IV Methylprednisolone | High-dose pulse steroids | First-line; may reduce seizures and slow progression |
| Long-term corticosteroids | Oral prednisolone | Maintenance; limited by side effects |
| IVIg | Immunomodulation | Short-term benefit; needs repeated dosing |
| Plasma exchange | Antibody removal | Adjunct, especially if antibodies present |
| Tacrolimus / cyclosporine | Calcineurin inhibitors (T-cell suppression) | Some benefit in case series |
| Rituximab | Anti-CD20, B-cell depletion | Case reports; may help in early/active disease |
| Natalizumab | Anti-α4-integrin; reduces lymphocyte trafficking | Emerging evidence; adult cases |
| Adalimumab | Anti-TNF | Limited case reports |
Immunotherapy can slow progression but rarely stops it completely and does not cure the disease.
As noted in Harrison's Principles of Internal Medicine (21st ed., p. 2817), patients with autoimmune/paraneoplastic encephalitis "usually respond to treatment of the tumor, if found, and immunotherapy (e.g., glucocorticoids, IVIg, plasma exchange, rituximab, or cyclophosphamide)" — principles that overlap with RE management.
Surgical Treatment (Definitive)
Hemispheric surgery is the only treatment that can render patients seizure-free:
| Procedure | Description | Seizure Freedom Rate |
|---|---|---|
| Anatomical hemispherectomy | Complete removal of affected hemisphere | ~70–80% |
| Functional hemispherectomy | Disconnection with partial resection (less blood loss) | ~65–75% |
| Hemispherotomy | Pure disconnection; minimal tissue removal | Similar to functional; preferred modern approach |
Timing of surgery is a critical decision:
- Earlier surgery → better seizure outcomes, but greater neurological deficit from the procedure
- Later surgery → more established pre-existing deficits, but hemisphere already compromised
- Dominant hemisphere involvement adds significant risk (aphasia, language transfer possible if < ~6 years old)
Corpus callosotomy is a palliative option when hemispheric surgery is deferred.
Prognosis
- Without surgery: ~70% of patients develop significant permanent hemiplegia, aphasia (if dominant hemisphere), and cognitive impairment
- With early hemispheric surgery: seizure freedom in ~65–80%; neurological deficits are present but stable
- The contralateral hemisphere can compensate for motor and language functions, especially in younger children (neuroplasticity)
- Adult-onset RE generally progresses more slowly but may be harder to treat
Key Points Summary
- RE is a rare, chronic, progressive, unihemispheric autoimmune encephalitis
- Hallmark: medically refractory focal epilepsy + epilepsia partialis continua + progressive hemiplegia
- Pathology: CD8+ T-cell-mediated neuronal destruction
- MRI: progressive unihemispheric cortical atrophy with T2 hyperintensity
- Immunotherapy slows progression; hemispheric surgery is the only curative option
- Early surgical intervention in appropriate candidates is recommended before severe deficits establish
This is a shared conversation. Sign in to Orris to start your own chat.