Sle explain as 10 mark question
Reading File
Finding Sources
Searching PubMed
"systemic lupus erythematosus"[MeSH Terms] AND "pathogenesis"
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a complete 10-mark answer on Systemic Lupus Erythematosus (SLE):
Systemic Lupus Erythematosus (SLE)
Definition
Systemic Lupus Erythematosus is a chronic, multisystem autoimmune disease characterized by the production of pathogenic autoantibodies (especially anti-double-stranded DNA and anti-Sm), immune complex deposition, complement activation, and widespread tissue inflammation. It predominantly affects women of childbearing age (female:male ratio ~9:1), with higher prevalence in African American, Asian, and Hispanic populations.
Etiology and Predisposing Factors
SLE results from an interplay of genetic, hormonal, and environmental factors:
- Genetic: Multiple HLA and non-HLA gene associations including IRF5, STAT4, PTPN22, BLK, FCGR2A, and complement genes (C1q, C4 deficiencies strongly predispose to SLE)
- Hormonal: Estrogens promote autoimmunity; disease worsens during pregnancy and with oral contraceptive use
- Environmental triggers: UV light (photosensitivity), drugs (hydralazine, procainamide, isoniazid - drug-induced lupus), infections (EBV), smoking
Pathogenesis
SLE is a type III hypersensitivity (immune complex-mediated) disease. Key steps:
- Loss of tolerance to self-antigens - particularly nuclear antigens (DNA, histones, ribonucleoproteins)
- Autoantibody formation - B cells produce ANA, anti-dsDNA, anti-Sm antibodies. The earliest antibodies are against RNA-binding proteins (Ro), followed by anti-dsDNA, then anti-Sm/RNP around the time of clinical diagnosis
- Immune complex deposition - Antigen-antibody complexes deposit in kidneys (glomeruli), skin, joints, blood vessels, and choroid plexus, activating complement (C3a, C5a) and recruiting neutrophils and macrophages
- Type I Interferon signature - Plasmacytoid dendritic cells and low-density granulocytes (LDGs) produce large amounts of type I IFN (IFN-α), which amplifies B cell activation, dendritic cell maturation, and breaks peripheral tolerance
- NETosis - LDGs from SLE patients form NET (Neutrophil Extracellular Traps) excessively; NETs contain oxidized nucleic acids, LL37, and modified self-proteins that stimulate type I IFN via the cGAS/STING pathway and activate the NLRP3 inflammasome, releasing IL-1 and IL-18 that perpetuate tissue damage
- Complement activation and organ damage - Immune complex deposits activate classical complement → chemotaxis of inflammatory cells → fibrinoid necrosis → end-organ damage
Goldman-Cecil Medicine, p.2812; Firestein & Kelley's Textbook of Rheumatology
Clinical Features
SLE is a relapsing-remitting, multisystem disease. Common manifestations (with approximate frequency):
| System | Feature | Frequency |
|---|---|---|
| Cutaneous | Malar (butterfly) rash, discoid lupus, photosensitivity, oral ulcers, alopecia | 88% |
| Musculoskeletal | Arthritis/arthralgias (non-erosive, migratory) | 76% |
| Neuropsychiatric | Cognitive dysfunction, seizures, psychosis, headache | 66% |
| Serositis | Pleuritis, pericarditis | 63% |
| Hematologic | Hemolytic anemia, leukopenia, thrombocytopenia | 57% |
| Vascular | Raynaud phenomenon, vasculitis | 44-43% |
| Renal | Lupus nephritis (proteinuria, hematuria, casts) | 31% |
| Cardiac | Libman-Sacks endocarditis (non-infective), premature atherosclerosis | 18% |
Goldman-Cecil Medicine, Table 245-1
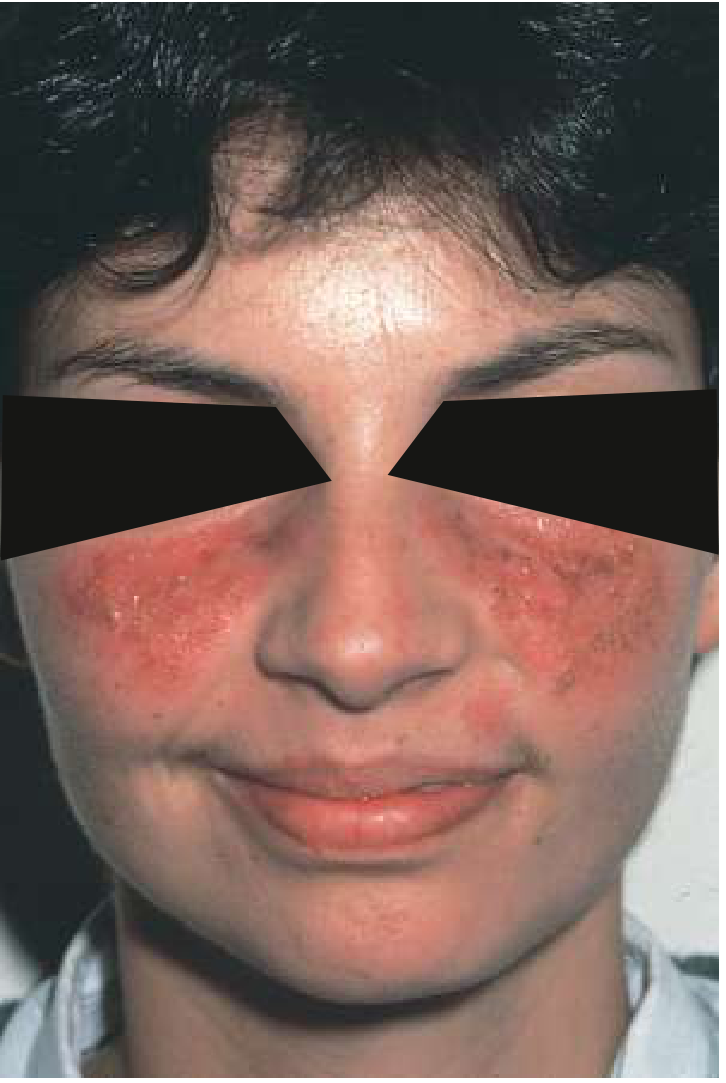
Malar Rash
The classic butterfly rash crosses both cheeks and the nasal bridge but characteristically spares the nasolabial folds:
Investigations
- ANA - Sensitive but not specific (positive in >95% of SLE; required entry criterion)
- Anti-dsDNA - Highly specific; titres correlate with disease activity (especially nephritis)
- Anti-Sm - Highly specific for SLE but less sensitive
- Complement levels - Low C3, C4 (consumed in active disease)
- Antiphospholipid antibodies (anticardiolipin, anti-β2GPI, lupus anticoagulant) - in ~30%; associated with thrombosis and pregnancy loss
- CBC - Anemia, lymphopenia, thrombocytopenia
- Urinalysis - Proteinuria, red cell casts in nephritis
- Renal biopsy - For lupus nephritis classification (WHO/ISN-RPS Class I-VI)
Diagnosis - 2019 ACR/EULAR Classification Criteria
SLE is classified when:
- ANA titer ≥ 1:80 (entry criterion - must be positive)
- Score ≥ 10 points from weighted clinical and immunologic domains
| Domain | Key Criterion | Points |
|---|---|---|
| Mucocutaneous | Acute cutaneous lupus | 6 |
| Musculoskeletal | Joint involvement | 6 |
| Immunologic | Anti-dsDNA antibody | 6 |
| Renal | Proteinuria >0.5g/24h | 4 |
| Complement | Low C3 AND C4 | 4 |
| Neuropsychiatric | Seizure | 5 |
| Constitutional | Fever | 2 |
Goldman-Cecil Medicine, Table 245
Management
General principles: Aim to suppress disease activity, prevent organ damage, and reduce cardiovascular risk.
- Hydroxychloroquine (HCQ) - Given to ALL SLE patients; reduces flares, thrombosis risk, and mortality. Mechanism: blocks TLR7/9 signaling
- NSAIDs / Low-dose corticosteroids - For mild disease (arthritis, serositis)
- Corticosteroids (moderate-high dose) - For major organ involvement (nephritis, CNS disease)
- Immunosuppressives:
- Mycophenolate mofetil (MMF) or cyclophosphamide - for lupus nephritis
- Azathioprine - maintenance therapy
- Methotrexate - for cutaneous/articular disease
- Biologics:
- Belimumab (anti-BAFF/BLyS) - approved for active SLE
- Anifrolumab (anti-IFN receptor) - for moderate-severe SLE
- Antiphospholipid antibody syndrome - Long-term anticoagulation
- Lifestyle: Sun protection, avoid triggers, vaccinations, manage CVD risk
Complications and Prognosis
- Lupus nephritis progresses to end-stage renal disease in ~10-20%
- Accelerated atherosclerosis and premature cardiovascular death
- Infections (major cause of early mortality due to immunosuppression)
- Avascular necrosis (from steroids or vasculitis)
- 10-year survival >90% with modern therapy; deaths in early disease are from organ failure/infection, later from cardiovascular disease
Summary: SLE is a prototype systemic autoimmune disease driven by failure of self-tolerance, autoantibody production (especially anti-dsDNA), immune complex deposition, and type I IFN dysregulation. It presents as a relapsing multisystem disorder diagnosed by positive ANA plus scored criteria. Treatment is layered from hydroxychloroquine (universal) to biologics for refractory disease.
Sources: Goldman-Cecil Medicine (International Edition); Firestein & Kelley's Textbook of Rheumatology (2022, Elsevier); Cellular and Molecular Immunology (9780323757485)
Create pdf of this
Reading File
Running Command
Running Command
Writing File
Running Command
Running Command
Your PDF is ready! Here is the download link:
SLE 10 Mark Answer
PDF Document · PDF
The PDF (375 KB) includes:
- 8 colour-coded sections with dark blue headers: Definition, Etiology, Pathogenesis, Clinical Features, Investigations, Diagnosis (2019 ACR/EULAR criteria), Management, and Complications/Prognosis
- Formatted tables for clinical manifestations, investigations, and classification criteria with alternating row colours
- Malar rash photograph with caption
- Summary box highlighting the key exam points
- Source citations at the bottom
Explain dumping syndrom like above
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Writing File
Running Command
Running Command
Here is the complete answer and the PDF:
Dumping Syndrome 10 Mark Answer
PDF Document · PDF
Dumping Syndrome
1. Definition
Dumping syndrome is a constellation of GI and vasomotor symptoms caused by rapid, unregulated emptying of gastric contents into the small intestine, bypassing normal pyloric regulation. It is classified into early dumping (10-30 min after meal) and late dumping (1-3 hours after meal).
2. Etiology and Predisposing Causes
Surgical (most common):
- Partial gastrectomy with Billroth II reconstruction - most common; loss of pyloric reservoir function
- Billroth I, Roux-en-Y reconstruction (less often)
- Vagotomy + pyloroplasty/gastroenterostomy - vagotomy abolishes receptive relaxation of proximal stomach, markedly accelerating liquid emptying
- Roux-en-Y gastric bypass (mild early dumping, usually self-limiting in 1-2 months)
- Oesophagectomy, Whipple procedure (non-pylorus sparing)
Non-surgical:
- Early type 2 diabetes (autonomic neuropathy), Zollinger-Ellison syndrome, idiopathic rapid gastric emptying
3. Pathophysiology
Early Dumping (osmotic + neurohormonal)
- Loss of pyloric function → hyperosmolar food bolus rapidly enters duodenum/jejunum
- Osmotic fluid shift from intravascular space into intestinal lumen → luminal distension + intravascular volume contraction
- Loss of vagally mediated receptive relaxation raises intragastric pressure, accelerating emptying further
- Duodenal bypass (Billroth II, Roux-en-Y) eliminates feedback receptors that normally slow emptying
- Rapid jejunal nutrient entry triggers release of vasoactive agents: neurotensin, VIP, GLP-1, serotonin, substance P
- Result: GI symptoms (pain, nausea, bloating, diarrhoea) + vasomotor symptoms (flushing, tachycardia, diaphoresis, syncope)
Late Dumping (reactive hypoglycaemia)
- Rapid carbohydrate delivery → quick absorption → hyperglycaemia
- Exaggerated GLP-1-mediated insulin release (overshoots) → reactive hypoglycaemia
- Hypoglycaemia activates adrenal glands → catecholamine release
- Result: diaphoresis, tremulousness, lightheadedness, tachycardia, confusion
4. Clinical Features
| Feature | Early Dumping (10-30 min) | Late Dumping (1-3 hours) |
|---|---|---|
| Mechanism | Osmotic + neurohormonal | Reactive hypoglycaemia |
| GI symptoms | Nausea, vomiting, pain, bloating, diarrhoea | Minimal |
| Vasomotor | Flushing, tachycardia, diaphoresis, syncope | Diaphoresis, tremor, weakness, confusion |
| Relief | Lying down for 30-60 min | Eating (raises blood glucose) |
| Frequency | ~75% of cases | ~25% of cases |
5. Diagnosis
- Primarily clinical - characteristic postprandial symptoms after appropriate gastric surgery
- Modified 75g OGTT:
- Early dumping positive: haematocrit rise ≥3% OR heart rate increase ≥10 bpm at 30 min
- Late dumping positive: blood glucose <50 mg/dL between 1-3 hours
- Radionuclide gastric emptying scintigraphy with a liquid marker (>30% emptied at 30 min = rapid); low sensitivity and specificity
- Continuous glucose monitoring (CGM): for late dumping hypoglycaemia episodes
6. Management
Step 1 - Dietary (first-line):
- Small, frequent meals (6/day); separate liquids from solids (30-45 min apart); avoid simple sugars; lie down after meals; add pectin/guar gum to retard emptying
Step 2 - Pharmacological (diet-refractory):
| Drug | Mechanism | Dose |
|---|---|---|
| Octreotide | Inhibits gastric emptying, slows bowel transit, inhibits GI hormones + insulin | 25-100 mcg SC before meals; depot 10-20 mg IM monthly |
| Acarbose | Blocks carb absorption, blunts glucose spike + hyperinsulinaemia | 50-100 mg TID with meals (late dumping) |
| GLP-1 receptor agonists | Delays gastric emptying, modulates insulin | Emerging therapy |
| Loperamide | Slows intestinal transit | For diarrhoea-predominant cases |
Step 3 - Surgery (refractory cases only, ~2% of patients):
- Billroth II → Roux-en-Y conversion (preferred)
- Pyloric reconstruction; takedown of gastrojejunostomy
7. Prognosis
Majority improve with diet alone. Persistent symptoms >2-3 months are uncommon and mainly seen after vagotomy. Good overall prognosis with stepwise management.
Sources: Sabiston Textbook of Surgery (2024); Fischer's Mastery of Surgery (8th ed.); Goldman-Cecil Medicine (2022); Sleisenger & Fordtran's GI and Liver Disease; Yamada's Textbook of Gastroenterology (7th ed.)
This is a shared conversation. Sign in to Orris to start your own chat.