Respiratory waves according to guyton n hall physiology book
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Here is the complete account of Respiratory Waves in the Arterial Pressure as presented in Guyton and Hall Textbook of Medical Physiology (Chapter 18, p. 232):
Respiratory Waves in the Arterial Pressure
What They Are
With each cycle of respiration, the arterial pressure rises and falls 4 to 6 mm Hg in a wave-like manner. During deep respiration, these swings can reach up to 20 mm Hg per respiratory cycle.
Mechanisms (3 Causes)
The waves result from several effects, some reflex in nature:
-
Spillover from the respiratory center to the vasomotor center
- Breathing signals arising in the medullary respiratory center spill over into the vasomotor center with each respiratory cycle, periodically altering vasomotor tone.
-
Intrathoracic pressure changes during inspiration
- During inspiration, intrathoracic pressure becomes more negative, causing thoracic blood vessels to expand.
- This reduces blood return to the left heart, which momentarily decreases cardiac output and arterial pressure.
-
Excitation of vascular and atrial stretch receptors
- Pressure changes in thoracic vessels during respiration can excite vascular and atrial stretch receptors, which reflexly influence vasomotor activity.
Net Result
- During early expiration: arterial pressure typically rises
- During the remainder of the respiratory cycle: pressure falls
Vasomotor Waves (Mayer Waves) - Related Concept
The book immediately contrasts respiratory waves with vasomotor waves (Mayer waves) - larger oscillations of 10 to 40 mm Hg that are slower than respiratory waves:
- Cycle duration: 26 seconds in anesthetized dogs; 7-10 seconds in unanesthetized humans
- These represent oscillations of reflex pressure control systems, not simple respiratory mechanics
Two causes of vasomotor waves:
1. Oscillation of Baroreceptor and Chemoreceptor Reflexes (Fig. 18.11B)
- High pressure excites baroreceptors -> sympathetic inhibition -> pressure falls a few seconds later
- Reduced pressure removes baroreceptor stimulation -> vasomotor center reactivates -> pressure rises again
- This delayed feedback loop creates cyclical oscillations
- The chemoreceptor reflex oscillates simultaneously; it dominates when pressure is in the 40-80 mm Hg range (where baroreceptor control is weaker)
2. Oscillation of the CNS Ischemic Response (Fig. 18.11A)
- When cerebrospinal fluid pressure is elevated (e.g., to 160 mm Hg), cerebral vessel compression triggers the CNS ischemic response, driving arterial pressure up to ~200 mm Hg
- Relief of ischemia at high pressure makes sympathetic activity fall -> pressure drops rapidly
- Brain ischemia recurs -> pressure rises again, and the cycle repeats
Key Principle
The book emphasizes that any reflex pressure control mechanism can oscillate if:
- The feedback intensity is strong enough, AND
- There is a delay between excitation of the pressure receptor and the subsequent pressure response
This is analogous to mechanical/electrical control systems - just as an automatic pilot with excessive feedback gain and response delay causes an aircraft to oscillate rather than fly straight.
Guyton and Hall Textbook of Medical Physiology, Chapter 18, p. 232
Give whole necroptosis pathway n what it is according to cell signalling
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I have everything needed. Here is the complete, detailed account:
Necroptosis - Complete Pathway & Cell Signalling
What is Necroptosis?
Necroptosis is a form of programmed cell death that morphologically resembles necrosis but is mechanistically controlled by specific intracellular signalling cascades - hence the name "programmed necrosis." It is distinct from both classical necrosis and apoptosis:
| Feature | Apoptosis | Classical Necrosis | Necroptosis |
|---|---|---|---|
| Controlled/programmed | Yes | No (passive/accidental) | Yes |
| Caspase-dependent | Yes | No | No (caspase-independent) |
| Plasma membrane rupture | No (membrane blebs) | Yes | Yes |
| Inflammation triggered | Minimal | Yes | Yes |
| ATP loss | No | Yes | Yes |
| ROS generation | Minimal | Yes | Yes |
| Lysosomal enzyme release | No | Yes | Yes |
The Complete Necroptosis Pathway
Step 1: Trigger Signals
Necroptosis is activated by multiple upstream stimuli:
- TNFR1 ligation (most studied model) - by TNF (Tumour Necrosis Factor)
- Fas/CD95 ligation
- Toll-like receptors (TLR3, TLR4) - pattern recognition in innate immunity
- T-cell receptor (TCR) signalling
- Viral DNA/RNA sensors - cytoplasmic sensors detect viral nucleic acids
- DNA damage
Critical condition: Necroptosis preferentially occurs when caspase-8 is inactive or inhibited. Normally, active caspase-8 cleaves RIPK1 and RIPK3, suppressing necroptosis in favour of apoptosis. When caspases are blocked (e.g., by viral caspase inhibitors or pharmacological agents), the necroptotic cascade is unleashed.
Step 2: RIPK1-RIPK3 Complex (Necrosome) Formation
The central signalling hub of necroptosis:
- TNFR1 is ligated by TNF (trimerized TNF binds to trimerized receptor)
- Ligation recruits RIPK1 (Receptor-Interacting Protein Kinase 1) into a multiprotein complex at the intracellular death domain of TNFR1
- RIPK1 is phosphorylated (auto- and trans-phosphorylation)
- Phosphorylated RIPK1 recruits and activates RIPK3 (Receptor-Interacting Protein Kinase 3)
- RIPK1 and RIPK3 form the necrosome - a multiprotein signalling complex
Note: RIPK1 is not always required - in some TLR3/4 and viral RNA-triggered necroptosis, RIPK3 can be activated independently of RIPK1.
Step 3: MLKL Phosphorylation (The Executioner Step)
This is the defining molecular event of necroptosis:
- Within the necrosome, RIPK3 phosphorylates MLKL (Mixed Lineage Kinase domain-Like protein) at key serine/threonine residues
- Phosphorylated MLKL undergoes a conformational change
- MLKL monomers assemble into oligomers (homo-oligomerization)
- MLKL oligomers translocate from the cytosol to the plasma membrane
- MLKL functions as an endogenous porin - it inserts into and disrupts the plasma membrane
Step 4: Plasma Membrane Disruption and Cell Death
- Plasma membrane rupture leads to cell swelling (oncosis) - microscopically visible as a "balloon-like structure"
- Cytoplasmic contents are released into the extracellular space
- This release includes DAMPs (Damage-Associated Molecular Patterns) - endogenous danger signals
- Result: Cell death + sterile inflammation
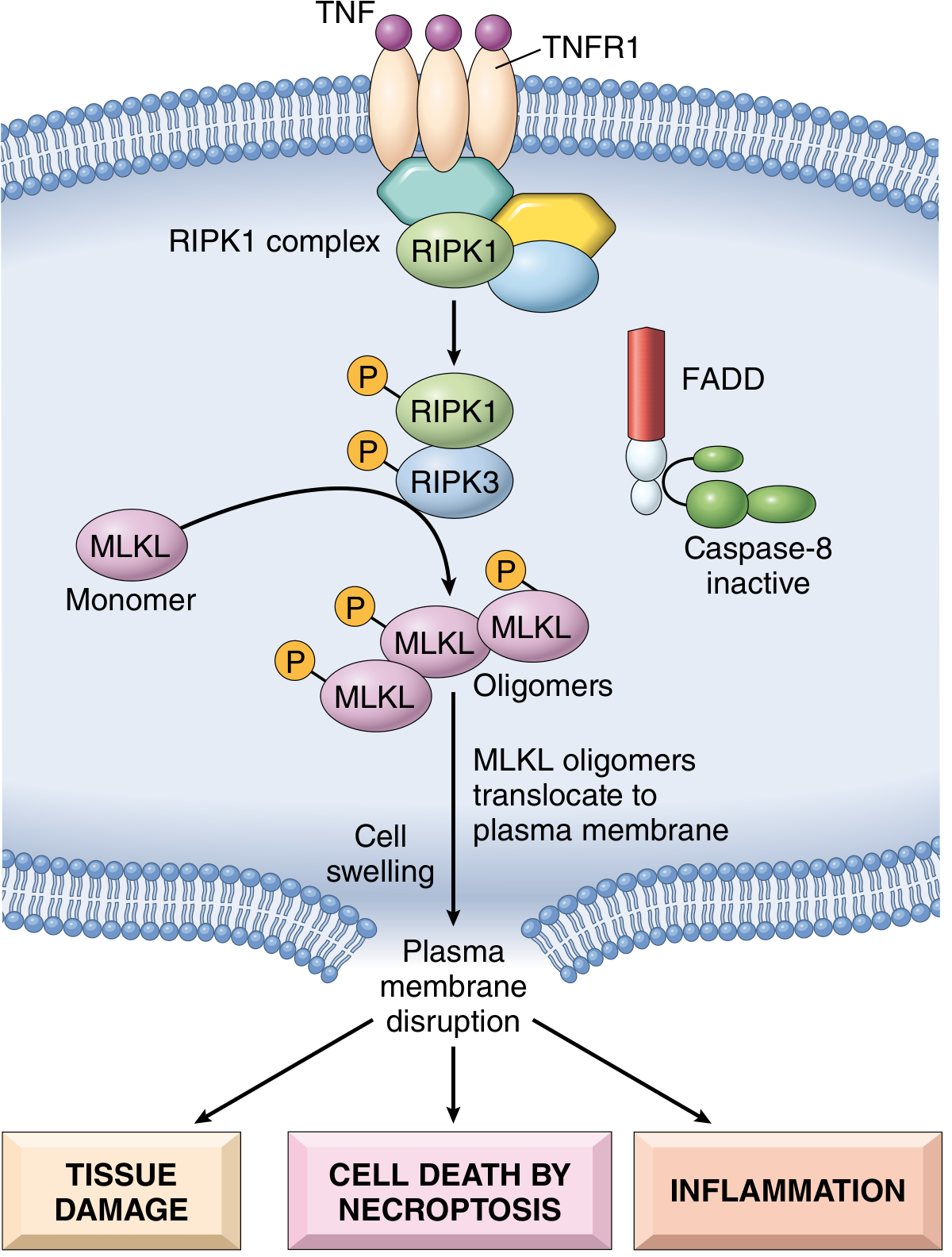
Fig. 2.17 - Molecular mechanism of TNF-mediated necroptosis (Robbins & Cotran)
Why Does Necroptosis Occur? (Caspase-8 as the Switch)
The decision between apoptosis and necroptosis is regulated by caspase-8 activity:
- Caspase-8 ACTIVE → cleaves RIPK1 and RIPK3 → necrosome cannot form → apoptosis proceeds
- Caspase-8 INACTIVE (e.g., viral inhibitors, pharmacological blockade) → RIPK1/RIPK3 remain intact → necroptosis proceeds
This makes necroptosis a backup death mechanism when viruses try to protect infected host cells by blocking apoptosis.
Downstream Consequences
The release of cellular contents triggers:
- Tissue damage - local destruction
- Inflammation - DAMP-mediated recruitment of innate immune cells; likely contributes to systemic inflammatory response
- Release of internalized pathogens into the extracellular space where complement and neutrophils can destroy them
ROS and Metabolic Collapse
In TNF/TNFR1-mediated necroptosis (especially when caspase-8 is pharmacologically inactivated), there is also:
- Over-generation of reactive oxygen species (ROS)
- Metabolic collapse (ATP depletion)
These further amplify the necrotic-like cell death.
Inhibitors
- Necrostatin-1: a specific RIPK1 inhibitor - the key research tool to confirm necroptosis is occurring
- RIPK3 knockout / MLKL knockout mice are used experimentally to study necroptosis
Physiological and Pathological Roles
| Context | Role |
|---|---|
| Bone growth plate formation | Physiological necroptosis drives terminal differentiation |
| Viral infection (e.g., CMV) | Backup killing when viruses inhibit caspases |
| Ischemia-reperfusion injury (brain, kidney, liver) | Pathological RIPK3/MLKL activation |
| Steatohepatitis | Contributes to hepatocyte death |
| Acute pancreatitis | Cell death mechanism |
| Neurodegenerative diseases (Parkinson's) | Chronic neuronal death |
| Cancer | Potential target when apoptosis resistance develops |
Key Summary (Robbins & Cotran)
"Necroptosis resembles necrosis morphologically, but like apoptosis is a genetically controlled form of cell death. Necroptosis is triggered by ligation of TNFR1 and by proteins found in RNA and DNA viruses. Necroptosis is caspase independent and depends on the RIPK1-RIPK3 complex. RIPK1-RIPK3 signaling leads to the phosphorylation of MLKL, which then forms pores in the plasma membrane. Release of cellular contents evokes an inflammatory reaction as in necrosis."
- Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 61
- Medical Physiology (Boron & Boulpaep)
- Scott-Brown's Otorhinolaryngology, Head & Neck Surgery Vol. 1
This is a shared conversation. Sign in to Orris to start your own chat.