serological test for hepatitis B
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have all the information needed for a comprehensive answer. Let me compile it.
Serological Tests for Hepatitis B
Hepatitis B diagnosis relies on a panel of antigens, antibodies, and molecular markers. Each has a distinct clinical meaning depending on whether infection is acute, chronic, resolved, or from vaccination.
The Key Markers
| Marker | Full Name | Clinical Meaning |
|---|---|---|
| HBsAg | Hepatitis B surface antigen | Infected (acute or chronic) and potentially infectious |
| Anti-HBs | Antibody to HBsAg | Immune (via recovery or vaccination) |
| Anti-HBc (total) | Antibody to hepatitis B core antigen | Exposed to HBV at some point |
| IgM anti-HBc | IgM class anti-HBc | Recent/acute infection (first antibody to appear) |
| IgG anti-HBc | IgG class anti-HBc | Past or chronic infection |
| HBeAg | Hepatitis B e antigen | High viral replication, high infectivity |
| Anti-HBe | Antibody to HBeAg | Generally low replication; but can be high in precore/BCP variants |
| HBV DNA | Viral nucleic acid | Direct indicator of active replication |
(Yamada's Textbook of Gastroenterology, 7th ed. - Table 87.2)
Individual Marker Details
HBsAg
- The principal screening test for HBV infection
- Appears approximately 1 month after infection (~4 weeks), detected as early as 1-2 weeks and as late as 11-12 weeks
- HBV DNA can actually be detected 2 weeks before HBsAg appears
- Persistence beyond 6 months = chronic infection
- Positive in both acute and chronic infection
Anti-HBs
- Indicates immunity - either from natural recovery or vaccination
- Appears after HBsAg disappears (following a gap called the "window period")
- The only anti-HBV antibody that is protective/neutralizing
- After vaccination: anti-HBs is present but anti-HBc is absent
Anti-HBc (IgM and IgG)
- IgM anti-HBc: The first antibody detected; present during acute infection; appears at onset of symptoms; may be the only marker detectable during the window period
- IgG anti-HBc: Replaces IgM with recovery; persists for life as a marker of prior exposure
- Anti-HBc is not neutralizing and does not confer immunity
- (Note: IgM anti-HBc may reappear during acute reactivation of chronic hepatitis B)
HBeAg
- Signifies active viral replication
- Presence = high HBV DNA levels (up to >10⁹ IU/mL)
- Absence + anti-HBe = lower replication (but can still be high in precore/BCP mutants)
Serological Course: Acute HBV Infection with Resolution
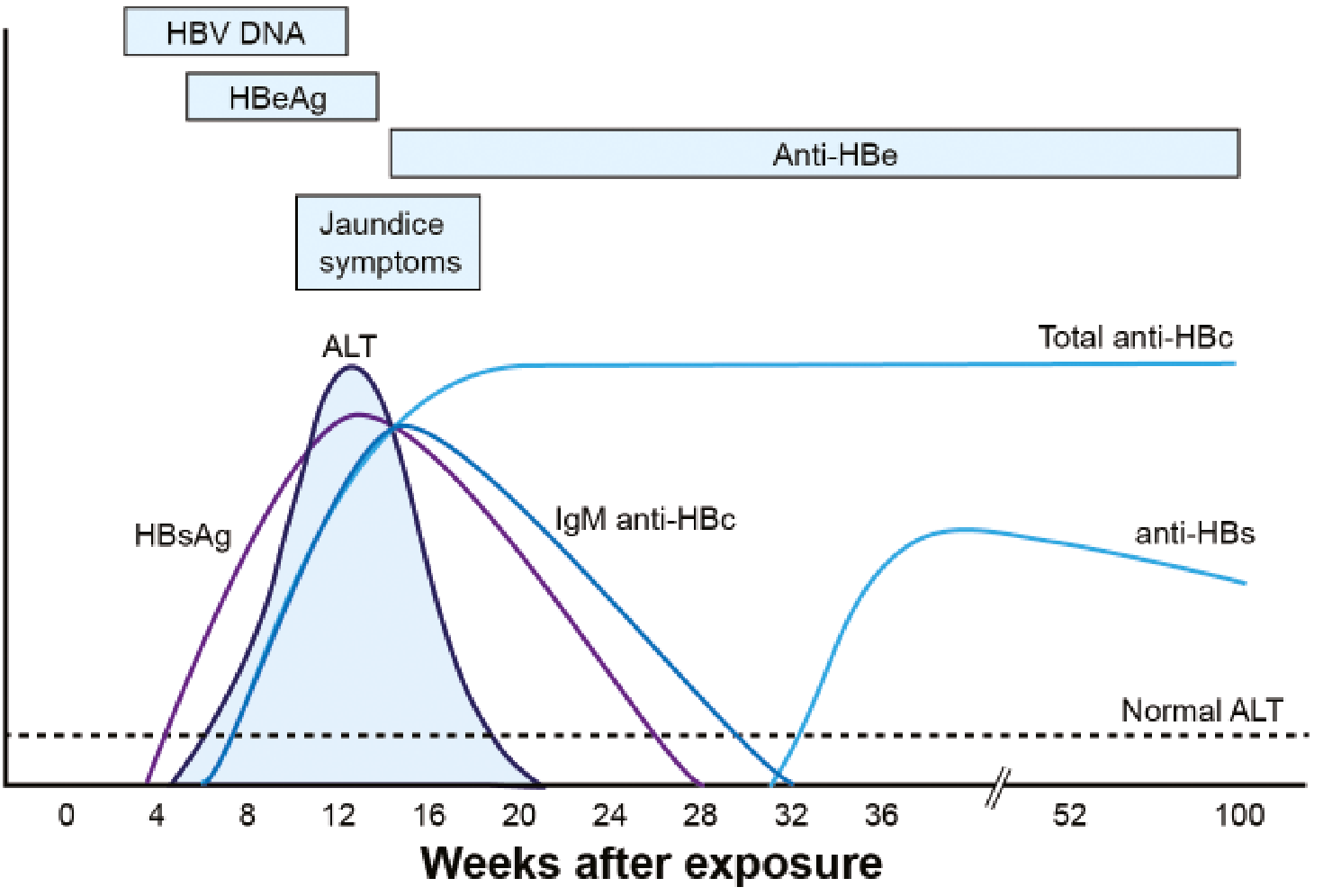
Key sequence in acute resolving infection:
- HBV DNA rises first (detectable ~2 weeks before HBsAg)
- HBsAg and HBeAg appear (weeks 4-8)
- ALT and jaundice peak after HBV DNA peaks (~week 12)
- IgM anti-HBc appears at symptom onset - may be the only marker in the window period
- HBsAg and HBeAg disappear (~week 20)
- Anti-HBe appears
- Window period: HBsAg gone, anti-HBs not yet present - only anti-HBc detectable
- Anti-HBs appears (~week 32+) - signals recovery and immunity
- IgM anti-HBc replaced by IgG anti-HBc (persists for life)
Serological Course: Progression to Chronic Infection
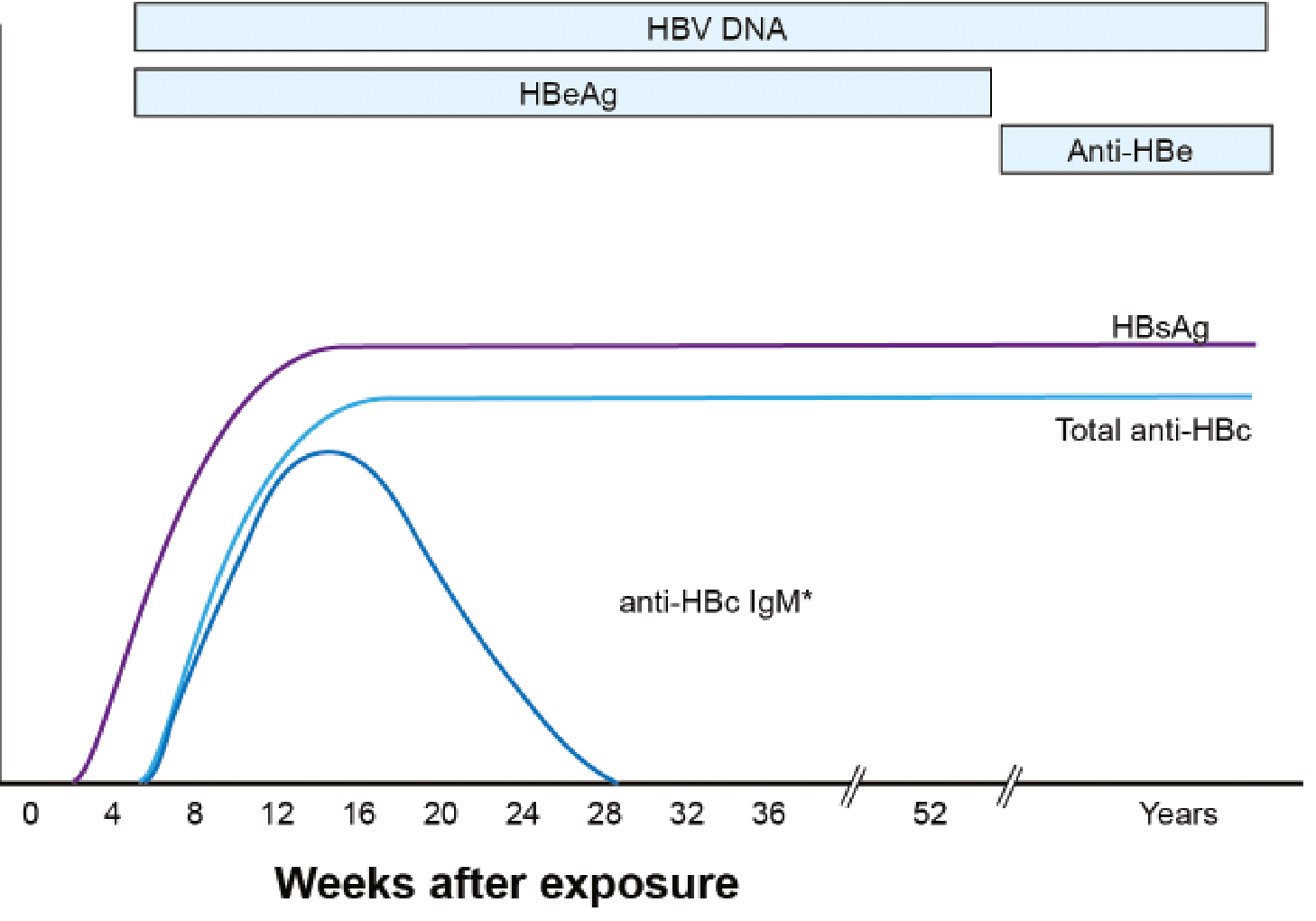
- HBsAg persists beyond 6 months
- HBV DNA remains detectable
- IgM anti-HBc wanes; total anti-HBc persists
- Anti-HBs never develops in chronic infection
Pattern-Based Interpretation Table
(From Harrison's Principles of Internal Medicine, 22nd ed., Table 350-5 and Jawetz Medical Microbiology)
| HBsAg | Anti-HBs | Anti-HBc | HBeAg | Anti-HBe | Interpretation |
|---|---|---|---|---|---|
| + | - | IgM | + | - | Acute hepatitis B, high infectivity |
| + | - | IgG | + | - | Chronic hepatitis B, high infectivity |
| + | - | IgG | - | + | Chronic/late acute HBV, low infectivity; or HBeAg-negative (precore mutant) |
| - | - | IgM | +/- | +/- | Window period in acute infection |
| - | - | IgG | - | +/- | Resolved infection (remote past) OR low-level carrier |
| - | + | IgG | - | - | Recovery from hepatitis B (naturally immune) |
| - | + | - | - | - | Vaccination only (no prior infection) |
| - | - | - | - | - | Never infected - susceptible |
Isolated Anti-HBc (Anti-HBc Alone)
When only anti-HBc is positive (HBsAg-, anti-HBs-), the interpretation is unclear and may represent:
- Resolved infection (most common) - anti-HBs has waned over time
- False-positive anti-HBc - patient is actually susceptible
- Low-level chronic carrier
- Resolving acute infection (window period)
Investigation: test IgM anti-HBc and HBV DNA to clarify.
HBV DNA (Virological Marker)
- Provides the most direct measure of viral replication
- Measured in IU/mL; levels fluctuate in chronic HBV
- Threshold of 2,000 IU/mL is the key cut-off used in most chronic HBV guidelines
- HCC risk increases dramatically when baseline HBV DNA exceeds ~2,000 IU/mL
- Serial testing is important for management decisions and treatment monitoring
Newer Markers
Quantitative HBsAg
- HBV produces >1,000-fold more surface proteins than intact virions
- Reflects transcriptional activity of covalently closed circular DNA (cccDNA)
- Cut-off of 1,000 IU/mL proposed to differentiate HBeAg-negative chronic hepatitis from inactive carrier state
- Better predictor of HCC and liver-related mortality than HBV DNA in HBeAg-negative patients with low viremia (<2,000 IU/mL)
Quick Summary: Clinical Status vs. Test Results
| Clinical Status | HBsAg | Anti-HBs | Anti-HBc (total) | IgM anti-HBc |
|---|---|---|---|---|
| Susceptible (never infected) | - | - | - | - |
| Acute infection | + | - | + | + |
| Window period | - | - | + | + |
| Resolved (immune, natural) | - | + | + | - |
| Vaccinated (immune) | - | + | - | - |
| Chronic infection | + | - | + | - |
(Quick Compendium of Clinical Pathology, 5th ed. - Section 13.9)
Sources: Yamada's Textbook of Gastroenterology (7th ed.), Harrison's Principles of Internal Medicine (22nd ed.), Jawetz Melnick & Adelberg's Medical Microbiology (28th ed.), Quick Compendium of Clinical Pathology (5th ed.)
pathogenesis if atherosclerosis according to Robbins
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
I now have everything needed. Here is the comprehensive answer directly from Robbins & Cotran Pathologic Basis of Disease.
Pathogenesis of Atherosclerosis (Robbins & Cotran)
The "Response-to-Injury" Hypothesis
Robbins frames atherosclerosis as a chronic inflammatory response of the vessel wall to endothelial injury - an attempt at "vascular healing" that goes wrong. The central concept is the response-to-injury hypothesis, which proposes that endothelial cell (EC) dysfunction is the initiating event.
Step-by-Step Pathogenesis
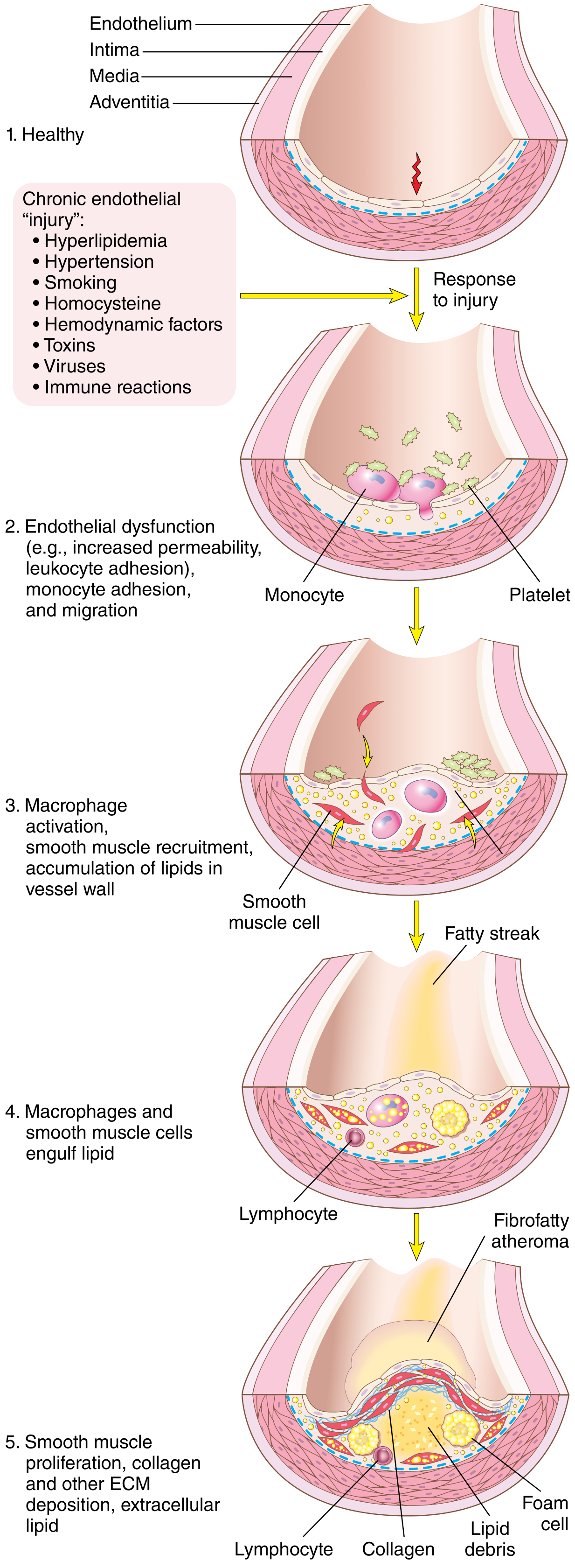
Step 1 - Endothelial Injury and Dysfunction
EC injury is the cornerstone of atherogenesis. Importantly, early lesions begin at sites of morphologically intact endothelium that shows functional abnormalities:
- Increased permeability
- Enhanced leukocyte adhesion
- Altered gene expression
Causes of endothelial dysfunction:
| Cause | Mechanism |
|---|---|
| Hemodynamic disturbances | Turbulent, non-laminar flow at branch points and ostia suppresses KLF2 (a transcription factor that drives atheroprotective genes) and activates pro-inflammatory gene transcription. Plaques therefore cluster at vessel ostia, branch points, and the posterior abdominal aorta. |
| Hypercholesterolemia | Directly impairs EC function; increases local reactive oxygen species (ROS); ROS accelerate NO decay, dampening vasodilation. |
| Hypertension, smoking, toxins, homocysteine | Additional causes of EC dysfunction listed in Robbins. |
| Inflammation / immune reactions | Trigger and amplify EC activation. |
Laminar flow normally upregulates Krüppel-like factor-2 (KLF2) which switches on atheroprotective genes. Statins also exert some of their benefit through KLF2 upregulation.
Step 2 - Lipid Accumulation (Hypercholesterolemia)
Evidence implicating hypercholesterolemia in atherogenesis (from Robbins):
- The dominant lipids in atheromatous plaques are cholesterol and cholesterol esters
- Genetic defects in lipoprotein uptake (e.g., familial hypercholesterolemia - loss-of-function LDL receptor mutations) cause accelerated atherosclerosis and MI before age 20 in homozygotes
- Epidemiologic correlation between total plasma cholesterol/LDL and atherosclerosis severity
- Lowering serum cholesterol slows progression, causes regression of some plaques, and reduces cardiovascular events
Mechanisms of lipid-driven atherogenesis:
- Chronic hyperlipidemia → lipoproteins accumulate in the intima
- Lipoproteins aggregate or become oxidized by free radicals from inflammatory cells
- Modified (oxidized) LDL is taken up by macrophages via scavenger receptors (not the normal LDL receptor)
- Because scavenger receptor-mediated uptake cannot be downregulated, macrophages become overloaded → foam cells (lipid-filled macrophages)
- Smooth muscle cells (SMCs) similarly become foam cells via LDL receptor-related proteins
- Modified lipoproteins are also directly toxic to ECs, SMCs, and macrophages
- This stimulates release of growth factors, cytokines, and chemokines → vicious cycle of inflammation
Step 3 - Inflammation and the Inflammasome
Chronic inflammation drives both initiation and progression of lesions.
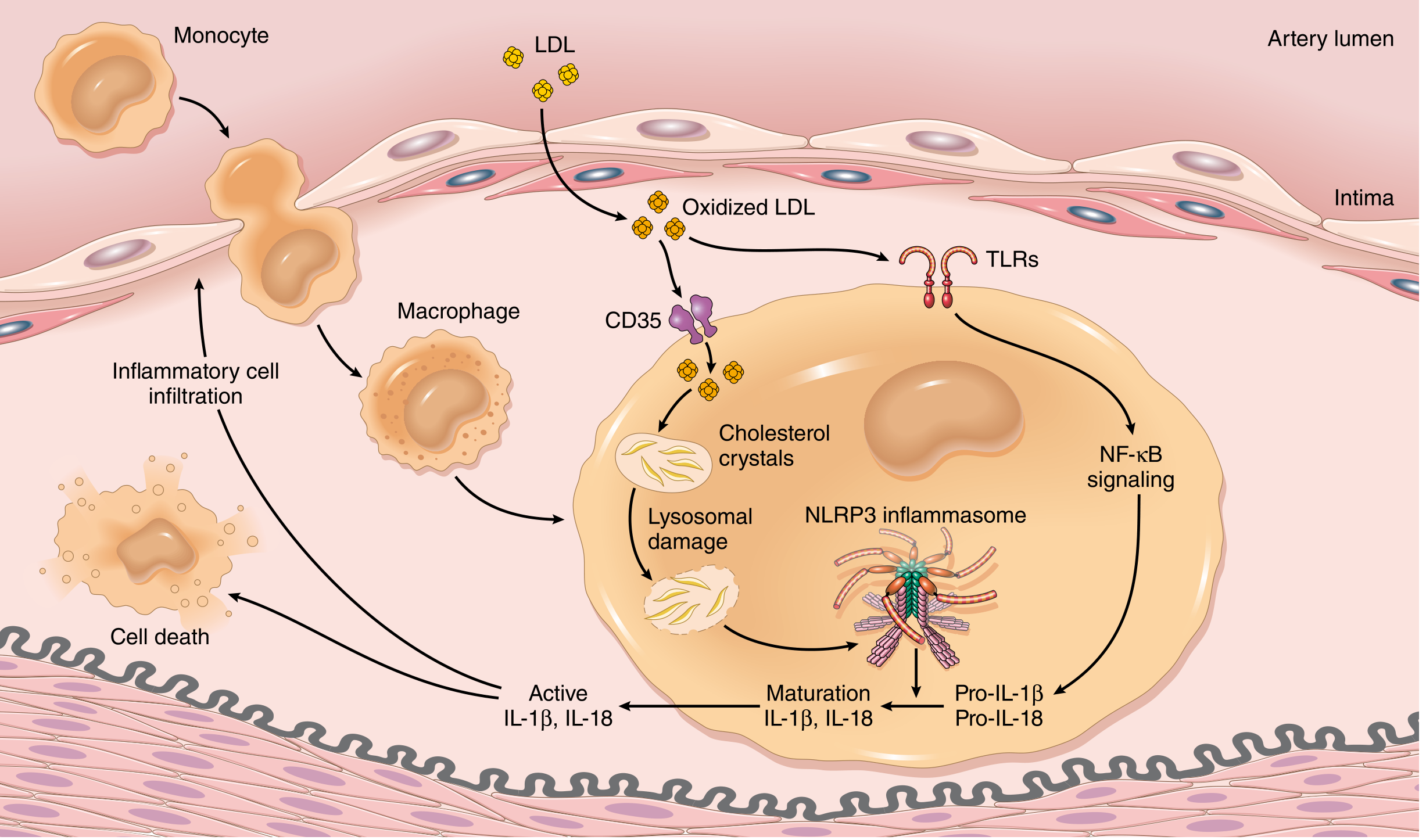
Key inflammatory sequence:
- Cholesterol crystals and free fatty acids accumulate in macrophages
- Sensed by cytosolic innate immune receptors → NLRP3 inflammasome assembly
- Inflammasome activation → production of IL-1β and IL-18
- IL-1 activates ECs and recruits/activates mononuclear cells (macrophages + T lymphocytes)
- Activated macrophages produce ROS → further LDL oxidation, plus growth factors driving SMC proliferation
- Activated T lymphocytes elaborate interferon-γ → activates macrophages, ECs, and SMCs
- This creates a self-amplifying loop of inflammatory cell recruitment and vessel wall injury
Step 4 - Monocyte Recruitment and Foam Cell Formation (Fatty Streak)
- Dysfunctional ECs express adhesion molecules (VCAM-1, ICAM-1, selectins) → monocytes and T lymphocytes adhere
- Monocytes migrate into the intima → differentiate into macrophages
- Macrophages and SMCs engulf oxidized LDL via scavenger receptors → transform into foam cells
- Foam cells accumulate in the intima → first visible lesion = fatty streak (appears as yellow streaks on intimal surface, near vessel ostia)
- Fatty streaks are present even in children and adolescents
Step 5 - Smooth Muscle Cell Proliferation and ECM Synthesis (Plaque Progression)
- Growth factors (PDGF from platelets, macrophages, ECs, SMCs; FGF; TGF-α) recruit medial SMCs into the intima
- SMCs proliferate and synthesize ECM proteins (especially collagen) → forms the fibrous cap
- This converts a fatty streak into a mature fibrofatty atheroma
- The fibrous cap stabilizes the plaque
- Conversely, activated inflammatory cells release matrix metalloproteinases (MMPs) that degrade ECM → destabilizes plaques → prone to rupture
Plaque Morphology (Mature Atheroma)
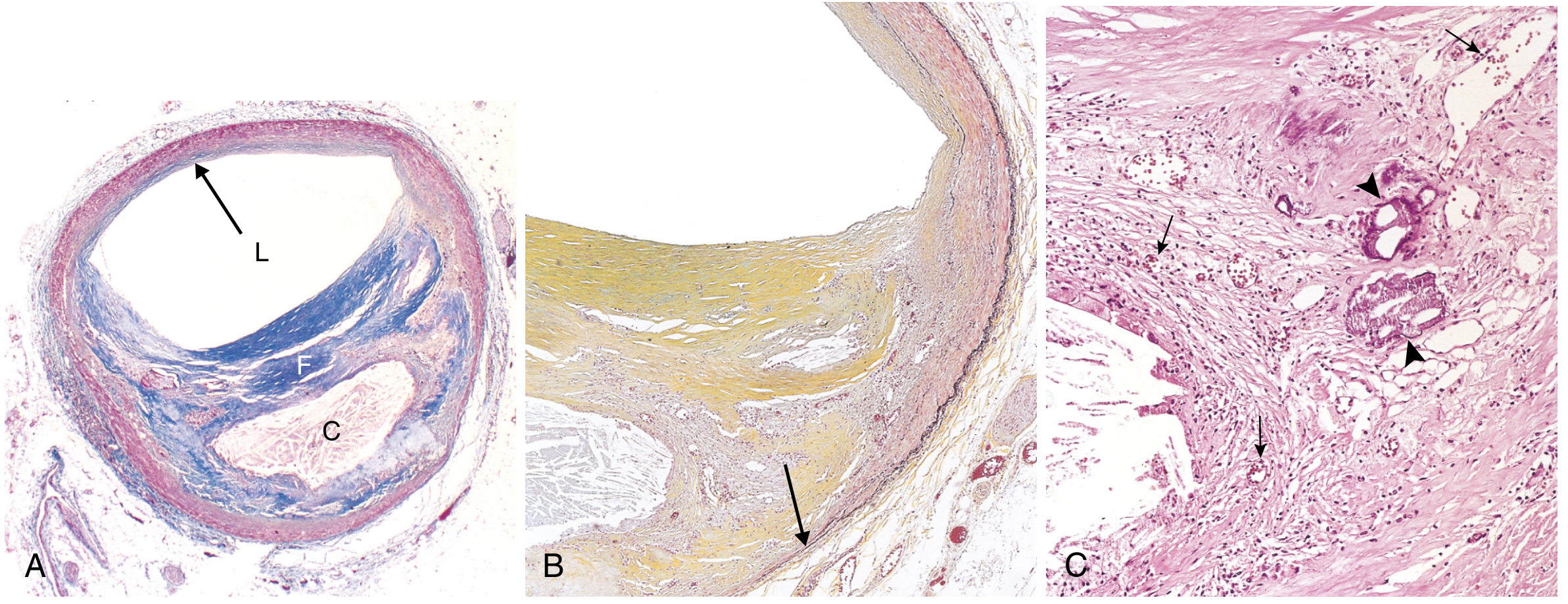
A fully developed atherosclerotic plaque has:
- Fibrous cap - SMCs embedded in dense collagen/ECM; surface covered by (often dysfunctional) ECs
- Necrotic/lipid core - extracellular lipid, cholesterol crystals, necrotic cell debris, foam cells
- Shoulders - region at plaque edges containing macrophages, T lymphocytes, SMCs
- Calcification - common in advanced plaques
- Neovascularization - new small vessels from adventitia (vasa vasorum) may supply the plaque
Stable vs. Vulnerable (Unstable) Plaques
| Feature | Stable Plaque | Vulnerable (Unstable) Plaque |
|---|---|---|
| Fibrous cap | Thick, dense collagen | Thin |
| Lipid core | Minimal | Large |
| Inflammation | Minimal | Dense macrophage/T-cell infiltrate |
| Risk | Chronic ischemia via luminal narrowing | Rupture, thrombosis, MI |
Plaque rupture exposes the subendothelial collagen and necrotic core → platelet aggregation + coagulation cascade → acute thrombus → sudden complete occlusion → myocardial infarction or stroke.
Consequences of Atherosclerosis
- Luminal narrowing - chronic ischemia (stable angina, claudication)
- Plaque rupture/thrombosis - acute MI, stroke (most feared)
- Atheroembolism - debris discharged into bloodstream causes microemboli
- Aneurysm formation - ischemic atrophy of media with elastic tissue loss → wall weakening → dilation/rupture
KEY CONCEPTS (Robbins Summary)
- Atherosclerosis is an intimal-based lesion with a fibrous cap and atheromatous core; constituents include SMCs, ECM, inflammatory cells, calcifications, lipids, and necrotic debris
- Atherogenesis is driven by interplay of vessel wall injury and inflammation
- All major risk factors converge on EC dysfunction and influence inflammatory cell and SMC recruitment
- Plaques develop slowly over decades; stable plaques cause chronic ischemia; unstable plaques cause acute catastrophic ischemic events
(Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 11)
can you teach me the morphology of squamous cell carcinoma nd how can i memorize it
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have everything - rich textbook content plus all the key images. Let me put together a detailed but student-friendly explanation with memory aids.
Morphology of Cutaneous Squamous Cell Carcinoma (SCC)
Let's break this down into Gross morphology → Microscopic morphology → Grading → Variants, with memory tricks at each step.
The Big Picture First
SCC arises from keratinocytes of the epidermis. Because these are cells that normally make keratin, the entire morphology story is about: how well can these cancer cells still make keratin?
Master memory peg: "SCC = Squamous = Keratin-making cells gone rogue. The better they still make keratin, the more well-differentiated they are."
GROSS MORPHOLOGY (What you see clinically)
SCC In Situ (Carcinoma in Situ / Bowen's disease)
- Sharply defined hyperkeratotic plaques or nodules
- Color ranges from erythematous to skin-colored
- Scaly, rough surface
- Often arise on top of prior actinic keratoses (sun-damaged precursors)
Invasive SCC
- Nodular, often scaly lesion
- Commonly ulcerates (classic "ulcerated nodule" appearance)
- Firm, indurated base
- Surrounding skin shows signs of sun damage (solar elastosis)
- Common sites: face, ears, lips, scalp, dorsum of hands - all sun-exposed
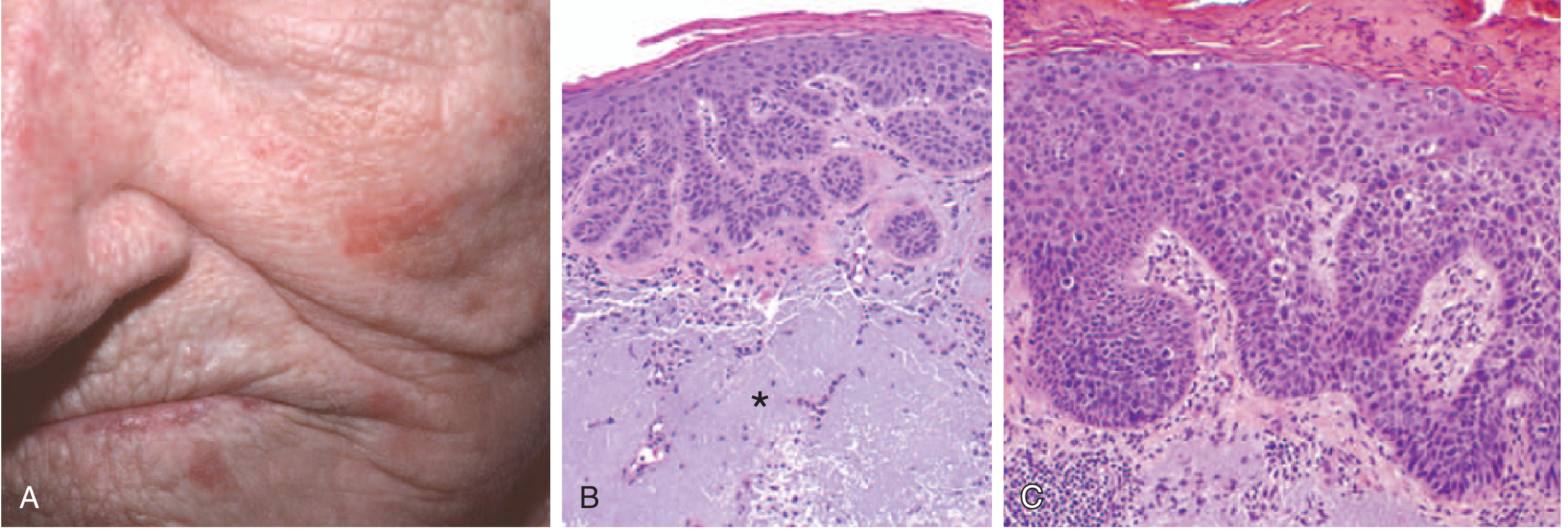
Memory trick for gross features: "NUSH" - Nodule → Ulcerates → Scaly surface → Hyperkeratotic (crusty top)
MICROSCOPIC MORPHOLOGY
The Defining Feature
The hallmark of invasive SCC is:
"Atypical keratinocytes originating in the epidermis and breaching the basement membrane to infiltrate the dermis."
This one sentence is the definition. Everything else describes how atypical those cells are.
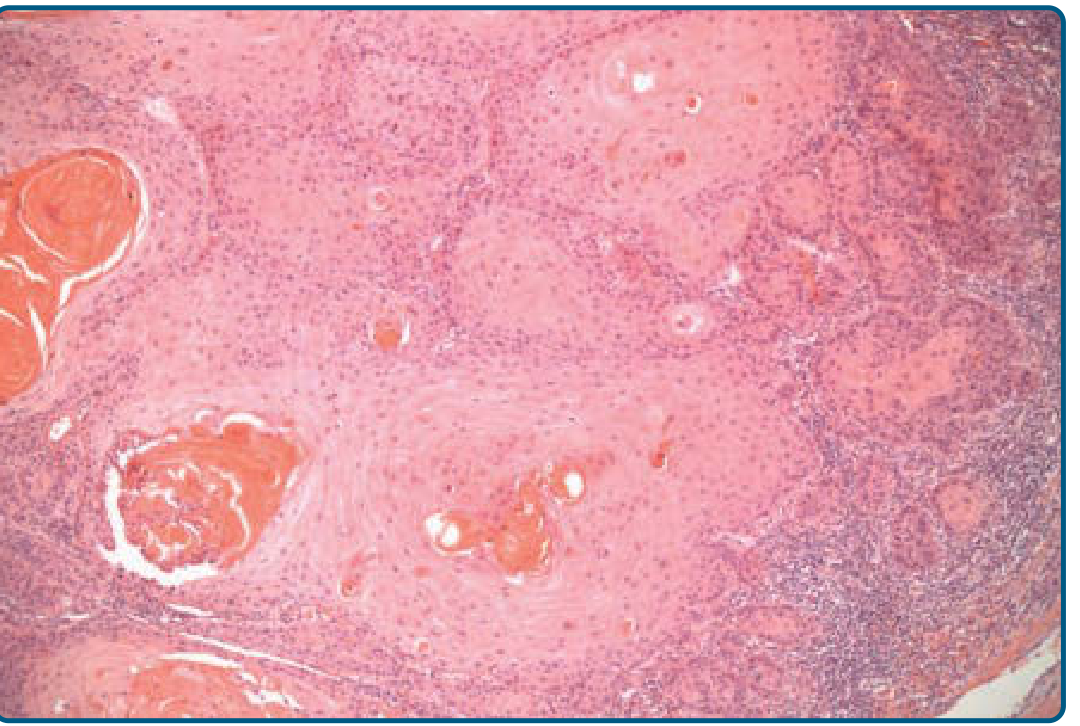
Key Microscopic Features (the "must-know" list)
| Feature | What it means | Well-differentiated | Poorly differentiated |
|---|---|---|---|
| Horn/Keratin pearls | Concentric whorls of keratin in the dermis - squamous cells wrapping around each other and keratinizing | Present, prominent | Absent or rare |
| Individual cell dyskeratosis | Single cells making keratin prematurely (before reaching surface) | Present | Less obvious |
| Parakeratosis | Nuclei retained in the stratum corneum | Present | Variable |
| Nuclear pleomorphism | Variation in nuclear size/shape | Minimal | Marked |
| Mitoses | Abnormal cell division figures | Few | Frequent, atypical |
| Intercellular bridges (prickles) | Visible desmosomes between cells on H&E | Usually visible | May be absent |
| Necrosis | Areas of dead tumor cells | Absent | Foci present |
THE GRADING SYSTEM (Broder's Grading)
The grade is determined by how well the cells still resemble normal squamous epithelium:
| Grade | Differentiation | Key Feature |
|---|---|---|
| Grade 1 | Well differentiated | Prominent keratinization, horn pearls, minimal pleomorphism |
| Grade 2 | Moderately differentiated | Intermediate features |
| Grade 3 | Poorly differentiated | Pleomorphic nuclei, frequent mitoses, very few areas of keratinization |
| Grade 4 | Undifferentiated/Anaplastic | Highly anaplastic cells, foci of necrosis, only abortive single-cell keratinization (dyskeratosis) |
Memory trick for grading: Think of it as "How much keratin is left?"
- Grade 1 = Lots of keratin (horn pearls everywhere) = "Keratinizing like crazy"
- Grade 4 = Almost no keratin (just single dying cells trying) = "Barely remembers it used to make keratin"
THE HORN PEARL - The Star Feature ⭐
This is the most asked-about and exam-favorite feature of SCC.
What is it?
A horn pearl (= keratin pearl / epithelial pearl) is a concentric, onion-skin arrangement of squamous cells with a central core of keratin, formed in the dermis.
Why does it form?
Because well-differentiated SCC cells still retain the property of keratinization - they pile up in nests and the central cells mature and produce keratin, forming a whorled "pearl."
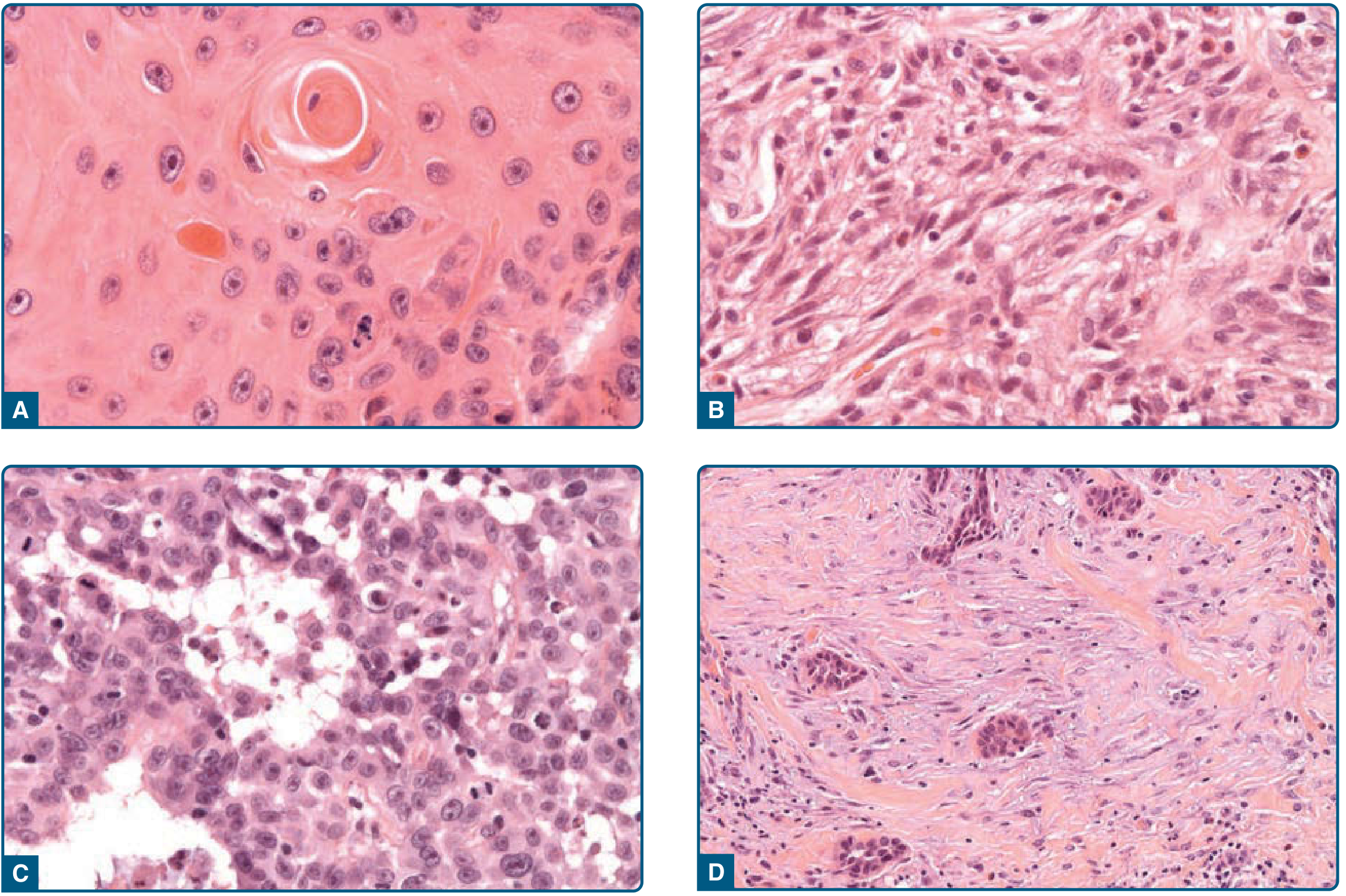
Memory trick: "A pearl forms by wrapping layers around a core - just like SCC cells wrap keratin around themselves in the dermis. The more pearl-forming, the more 'precious' (well-differentiated) the tumor."
HISTOLOGIC VARIANTS (High-Yield)
| Variant | Key Histologic Feature | Clinical Note |
|---|---|---|
| Common (typical) SCC | Atypical keratinocytes, horn pearls, invades dermis | Most frequent |
| Acantholytic (Adenoid) SCC | Acantholysis of atypical keratinocytes forming pseudoglandular spaces (looks like glands!) | <5% of SCCs; higher metastatic risk (~19%) |
| Spindle-cell SCC | Spindle-shaped atypical keratinocytes, no keratinization | Mimics sarcoma/melanoma; needs cytokeratin IHC to confirm |
| Verrucous SCC | Well-differentiated; broad, pushing borders; NO horn pearls, NO dyskeratosis | Slow-growing, locally destructive, low metastatic potential |
| Desmoplastic SCC | Infiltrative growth in abundant mucinous stroma | Highly infiltrative; high-risk variant |
Memory trick for variants - "A SAD V":
- A = Acantholytic (fake glands)
- S = Spindle cell (looks like sarcoma)
- A = Anaplastic/undifferentiated
- D = Desmoplastic (lots of stroma)
- V = Verrucous (wart-like, no pearls)
THE COMPLETE MICROSCOPIC PICTURE - One Paragraph to Memorize
"SCC in situ shows highly atypical cells at ALL levels of the epidermis with nuclear crowding and disorganization - but still above the basement membrane. Invasive SCC shows atypical keratinocytes breaching the basement membrane and infiltrating the dermis in nests or cords. Well-differentiated tumors form prominent horn pearls with individual cell dyskeratosis and parakeratosis. Poorly differentiated tumors show marked nuclear pleomorphism, frequent mitoses, foci of necrosis, and only abortive single-cell keratinization."
(Robbins & Kumar Basic Pathology + Fitzpatrick's Dermatology)
MASTER MEMORY FRAMEWORK
Here's the single mnemonic that covers everything:
"KAPID" for the 5 core micro features:
- K = Keratin pearls (horn pearls) - the signature
- A = Atypical keratinocytes (pleomorphic nuclei, mitoses)
- P = Parakeratosis + individual cell dyskeratosis
- I = Invasion through basement membrane into dermis
- D = Differentiation determines grade (lots of K → Grade 1; no K → Grade 4)
CLINICAL BEHAVIOR POINTS (for context)
- ~4% have already metastasized to regional lymph nodes at diagnosis
- Higher metastatic risk: lesions >2 cm, deep invasion below subcutaneous fat, perineural invasion, lesions near ear or lip
- SCC from actinic keratoses: locally aggressive but slow to metastasize
- SCC from burn scars, chronic ulcers, or non-sun-exposed skin: more aggressive
- Metastatic cutaneous SCC responds well to immune checkpoint inhibitors (due to high mutational burden from UV damage)
(Robbins & Kumar Basic Pathology, Chapter 22; Fitzpatrick's Dermatology, Chapter 112)
This is a shared conversation. Sign in to Orris to start your own chat.