Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a thorough, well-sourced explanation. Here is the complete answer:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Overview
ARDS is an acute, diffuse, inflammatory lung injury characterized by increased alveolar-capillary permeability, non-cardiogenic pulmonary edema, and severe hypoxemia. Unlike cardiogenic pulmonary edema (caused by elevated left-sided cardiac pressures), the edema in ARDS is exudative - meaning the alveolar-capillary barrier has been breached, allowing protein-rich fluid to flood the airspaces. The result is decreased lung compliance, profound hypoxemia, and increased dead space ventilation.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3145
Precipitating Causes
ARDS arises from either direct or indirect lung injury:
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia | Sepsis (most common) |
| Aspiration of gastric contents | Severe trauma / multiple fractures |
| Pulmonary contusion | Pancreatitis |
| Near-drowning | Multiple blood transfusions (TRALI) |
| Toxic inhalation | Burns, drug overdose |
- Harrison's Principles of Internal Medicine 22E, p. 2342
The Three-Phase Pathological Sequence

Figure: Time course of ARDS development, from Harrison's Principles
Phase 1 - Exudative (Days 0-7)
This is the acute injury phase and contains the core mechanism:
Step 1: Alveolar-capillary membrane disruption
The trigger (sepsis, pneumonia, trauma, etc.) initiates a systemic inflammatory response. Both the pulmonary microvascular endothelium and the alveolar epithelium (especially type I pneumocytes) are injured. Damage to the epithelium disrupts the normally tight barrier to fluid and macromolecules. Loss of pulmonary vascular endothelial barrier integrity is both necessary and sufficient for ARDS to develop.
Step 2: Protein-rich edema accumulates
With barrier breakdown, fluid rich in protein floods the interstitium and alveolar spaces. This exudate contains fibrin, fibrinogen, plasma proteins, and cellular debris. These proteins further inhibit surfactant function (see below).
Step 3: Neutrophil sequestration and activation (central mechanism)
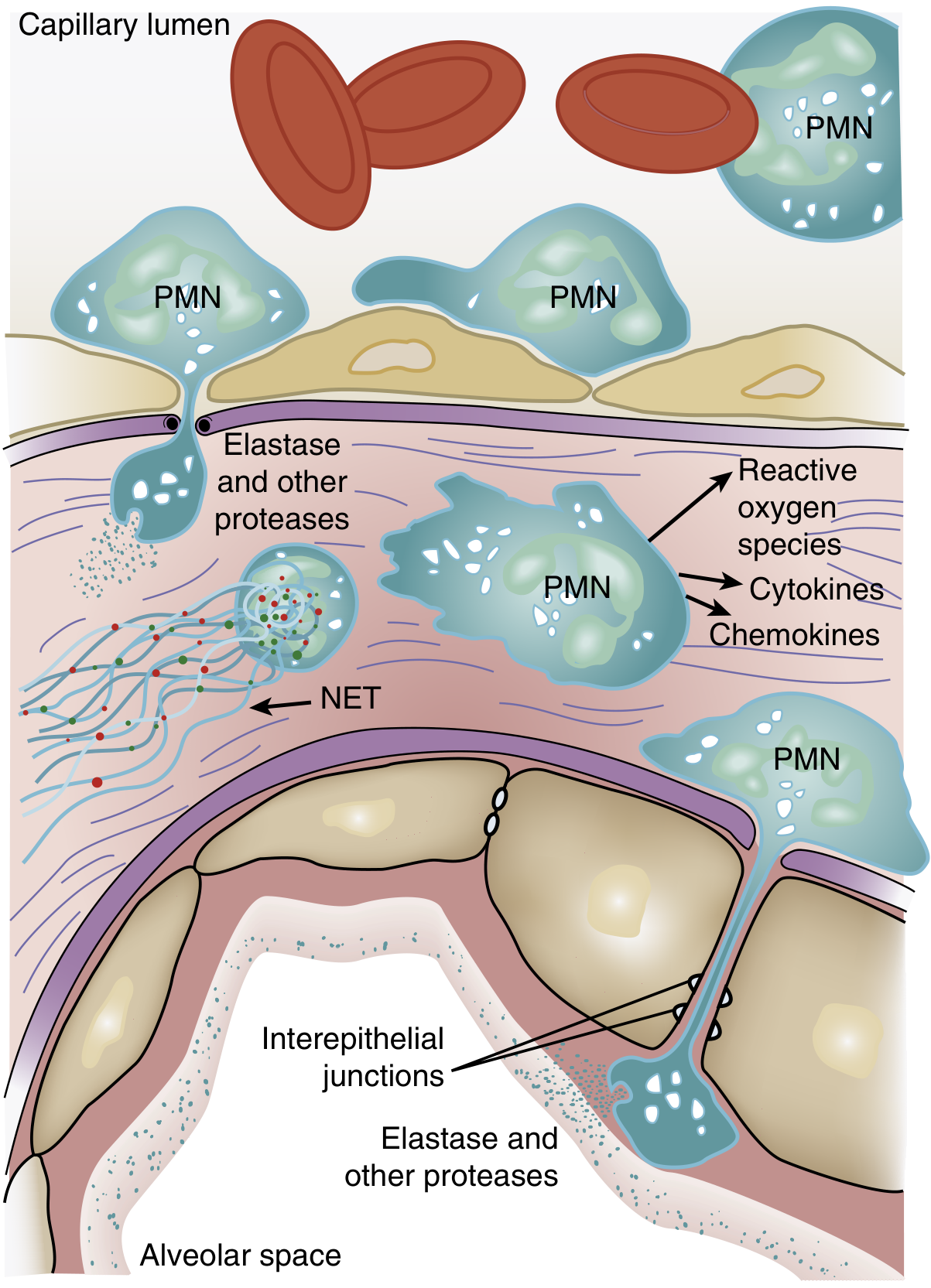
Figure: PMN-mediated injury to the alveolar-capillary unit, from Murray & Nadel's
One of the earliest events - often preceding visible hypoxemia - is a transient leukopenia due to massive neutrophil sequestration in the pulmonary microvasculature. This happens because:
- Pulmonary capillary diameter is smaller than average neutrophil diameter
- Activated neutrophils become "stiff" (via actin cytoskeleton changes) and cannot deform to pass through
- Sequestered neutrophils then migrate into the interstitium and alveolar space
Once in the lung, activated PMNs release a cascade of cytotoxic compounds:
- Reactive oxygen species (ROS) - directly damage cell membranes and proteins
- Proteolytic enzymes (leukocyte elastase, matrix metalloproteinases) - degrade structural proteins, break tight junctions, and degrade surfactant apoprotein A
- Cationic peptides (defensins)
- Cytokines (TNF-α, IL-1β) - amplify the inflammatory response
- Neutrophil extracellular traps (NETs) - contribute to tissue damage and microvascular occlusion
- Eicosanoids and platelet-activating factor - increase vascular permeability
Key signal transduction: p38 mitogen-activated protein kinase (MAPK) is activated by LPS and similar stimuli, driving TNF-α production and release of macrophage inflammatory protein-2 (a neutrophil chemokine), creating a positive feedback loop.
Step 4: Surfactant dysfunction
Type II pneumocytes are also injured, reducing surfactant production. The surfactant that remains is functionally impaired because:
- The proportion of large (active) to small (inactive) surfactant aggregates is reduced
- Plasma proteins (fibrin, albumin) that leaked in physically inhibit surfactant function
- Neutrophil elastase degrades surfactant protein A
- The composition of surfactant phospholipids (dipalmitoylphosphatidylcholine, phosphatidylglycerol) is altered
Loss of surfactant causes alveolar collapse (atelectasis) and dramatically worsens hypoxemia.
Step 5: Diffuse alveolar damage (DAD)
The combination of protein-rich edema, hemorrhage, hyaline membrane formation (from precipitated plasma proteins, fibrin, and necrotic cell debris), and neutrophilic infiltration is pathologically termed diffuse alveolar damage (DAD) - the morphologic hallmark of ARDS. Hyaline membranes line the alveolar ducts and respiratory bronchioles. About 50% of ARDS patients show DAD on autopsy; those with DAD have lower compliance, worse P/F ratios, and higher mortality.
Step 6: Physiological consequences
-
Hypoxemia: Protein-rich edema and atelectasis create massive intrapulmonary shunting (blood perfuses unventilated alveoli). Hypoxemia is refractory to supplemental oxygen alone.
-
Hypercapnia / increased dead space: Microvascular occlusion (from fibrin deposition, vasoconstriction from hypoxia, and vessel compression by positive-pressure ventilation) reduces perfusion to ventilated lung units, increasing dead space. This explains why minute ventilation rises to 12-16 L/min at onset.
-
Pulmonary hypertension: Due to hypoxic vasoconstriction, intravascular fibrin, and mechanical compression from ventilator pressures.
-
Decreased lung compliance: Alveolar flooding, atelectasis, and loss of surfactant result in a "stiff" lung. Gravity causes dependent consolidation, while non-dependent zones may be relatively preserved (heterogeneous involvement).
-
Murray & Nadel's Textbook of Respiratory Medicine, pp. 3145-3149
-
Harrison's Principles of Internal Medicine 22E, pp. 2343-2344
Phase 2 - Proliferative (Days 7-21)
In patients who survive the exudative phase, a repair response begins:
-
Hyaline membranes are reorganized
-
Interstitial inflammation becomes prominent
-
Type II pneumocytes proliferate to repopulate the denuded alveolar surface
-
Early fibrotic changes appear - elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid as early as 24 hours after onset, suggesting fibroproliferation may begin simultaneously with the inflammatory injury, not after it
-
Many patients recover during this phase, though dyspnea and hypoxemia may persist
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3074
Phase 3 - Fibrotic (>Day 21)
A subset of patients with persistent ARDS (>2 weeks) enter a fibrotic phase with:
- Established pulmonary fibrosis
- Obliteration of pulmonary capillaries
- Interstitial and alveolar collagen deposition
- Bulla formation
- Severe, often irreversible impairment of gas exchange
BAL fluid from ARDS patients stimulates cultured fibroblasts to proliferate in vitro, supporting active fibroproliferation. This phase carries high mortality.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3074-3076
Summary of Key Mechanisms
Trigger (sepsis, pneumonia, trauma)
↓
Systemic inflammatory response
↓
Neutrophil activation & sequestration in pulmonary capillaries
↓
PMN transmigration → ROS, elastase, cytokines (TNF-α, IL-1β), NETs
↓
Alveolar-capillary barrier disruption (endothelium + type I pneumocytes)
↓
Protein-rich exudate floods alveoli → hyaline membrane formation (DAD)
↓
Type II pneumocyte injury → surfactant loss → atelectasis
↓
V/Q mismatch + shunting → hypoxemia
Microvascular occlusion → increased dead space → hypercapnia
Pulmonary vasoconstriction → pulmonary hypertension
↓
Decreased compliance, respiratory failure
Berlin Definition (Diagnostic Criteria)
| Criterion | Specification |
|---|---|
| Timing | Acute onset within 1 week of clinical insult |
| Imaging | Bilateral opacities on CXR/CT not fully explained by effusions or collapse |
| Origin of edema | Not fully explained by heart failure or fluid overload |
| Oxygenation | Mild: PaO₂/FiO₂ 200-300 mmHg; Moderate: 100-200; Severe: <100 mmHg (on PEEP ≥5 cmH₂O) |
- Harrison's Principles of Internal Medicine 22E, p. 2343
This is a shared conversation. Sign in to Orris to start your own chat.