Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the necessary information to write a comprehensive answer. Let me compile everything.
Myocardial Infarction - Complete Review
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 joint task force of U.S. and European Cardiology groups defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
Classification
By ECG Pattern (Clinically Most Used)
| Type | ECG | Pathology |
|---|---|---|
| STEMI (ST-elevation MI) | ST segment elevation | Transmural infarct; complete coronary occlusion |
| NSTEMI (Non-ST-elevation MI) | No ST elevation | Subendocardial / partial thickness; incomplete occlusion |
| Unstable Angina | May be normal | No biomarker rise; ischemia without necrosis |
STEMI is invariably due to complete coronary artery occlusion and requires urgent thrombolysis or stenting. NSTEMI can often be managed conservatively.
By Depth
- Transmural MI - full thickness of myocardial wall involved; corresponds to STEMI
- Subendocardial MI - limited to inner 1/3 to 1/2 of myocardial wall; corresponds to NSTEMI
Fourth Universal Definition (Type Classification)
| Type | Cause |
|---|---|
| Type 1 | Spontaneous MI from atherothrombosis (plaque rupture/erosion) |
| Type 2 | MI due to supply-demand mismatch (e.g., vasospasm, tachycardia, anemia) |
| Type 3 | MI causing death before biomarkers can be confirmed |
| Type 4a/b | Percutaneous coronary intervention (PCI)-related MI |
| Type 5 | CABG surgery-related MI |
By Territory (based on coronary artery affected)
- LAD occlusion - anterior, anteroseptal, anterolateral wall of LV (most common; "widow maker")
- RCA occlusion - posterior, inferior wall of LV; right ventricle
- LCx occlusion - lateral wall of LV
Etiopathogenesis
Risk Factors
- Non-modifiable: Age, male sex, family history; approximately 10% of MIs occur below age 40
- Modifiable: Hypertension, hyperlipidemia, diabetes, smoking, obesity, sedentary lifestyle
- Women are protected during reproductive years due to estrogen; risk rises post-menopause
Pathogenesis Sequence (Atherothrombotic, ~90% of cases)
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and activate - releasing thromboxane A2, ADP, and serotonin, causing further aggregation and vasospasm
- Coagulation is activated by tissue factor exposure, adding to the growing thrombus
- Within minutes, the enlarging thrombus completely occludes the coronary lumen
Angiography within 4 hours of MI onset shows coronary thrombosis in ~90% of cases.
Non-atherothrombotic Causes (~10%)
- Coronary artery vasospasm (cocaine, ephedrine use)
- Embolism from mural thrombus (atrial fibrillation), valvular vegetations, paradoxical embolism
- Vasculitis, amyloid deposition, sickle cell disease (small vessel occlusion)
Myocardial Response to Ischemia
| Time | Event |
|---|---|
| Seconds | ATP depletion begins; aerobic metabolism ceases |
| < 2 min | Loss of contractility |
| 10 min | ATP reduced to 50% of normal |
| 20-40 min | Irreversible cell injury begins (necrosis threshold) |
| 40 min | ATP reduced to 10% of normal |
| > 1 hour | Microvascular injury |
| 6-12 hours | Progressive necrosis becomes complete |
The critical window is 20-40 minutes - severe ischemia lasting beyond this leads to irreversible myocyte death. The rationale for rapid reperfusion is to salvage as much at-risk myocardium as possible ("time is myocardium").
Necrosis begins in the subendocardial zone (most distal from epicardial vessels, exposed to high intramural pressures) and spreads as a "wavefront" toward the epicardium with prolonged ischemia.
Pathology: Morphology / Sequential Changes Over Time
(Robbins & Kumar Basic Pathology)
| Time Frame | Gross Appearance | Light Microscopy | EM Findings |
|---|---|---|---|
| 0-½ hr | None | None | Myofibril relaxation; glycogen loss; mitochondrial swelling |
| ½-4 hr | None | Usually none; wavy fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Occasionally dark mottling | Onset of coagulation necrosis; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Ongoing coagulation necrosis; pyknotic nuclei; hypereosinophilic myocytes; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan infarct center | Coagulation necrosis with loss of nuclei and striations; increased neutrophils | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Neutrophil disintegration; early macrophage phagocytosis at border | - |
| 7-10 days | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed macrophage phagocytosis; early granulation tissue at margins | - |
| 10-14 days | Red-gray depressed infarct borders | Well-established granulation tissue with new vessels and collagen deposition | - |
| 2-8 weeks | Gray-white scar, progressing from border to core | Increased collagen deposition; decreased cellularity | - |
| > 2 months | Scarring complete | Dense collagenous scar | - |
Key Microscopic Features
- Wavy fibers (½-4 hrs): stretching and buckling of non-contractile dead fibers at infarct edges
- Coagulation necrosis: preservation of cell outlines but loss of nuclei (4-12 hrs onward)
- Contraction band necrosis: particularly seen in reperfused infarcts - intense eosinophilic transverse bands formed by hypercontracted sarcomeres due to calcium influx
- Neutrophil infiltration: peaks 1-3 days
- Macrophage phagocytosis: peaks 5-10 days
- Granulation tissue: appears 1-2 weeks
- Dense collagenous scar: complete by 6 weeks
Reperfusion Injury
Restoring blood flow to ischemic tissue can cause additional damage. Mechanisms include:
- Mitochondrial dysfunction (membrane permeability → swelling → apoptosis)
- Myocyte hypercontracture (calcium overload)
- Free radical generation (superoxide, H2O2, peroxynitrite)
- Leukocyte aggregation causing microvascular "no-reflow" phenomenon
Laboratory Diagnosis (Cardiac Biomarkers)
The basis is measurement of intracellular proteins that leak from necrotic myocytes through damaged sarcolemmal membranes into the circulation.
Biomarker Kinetics
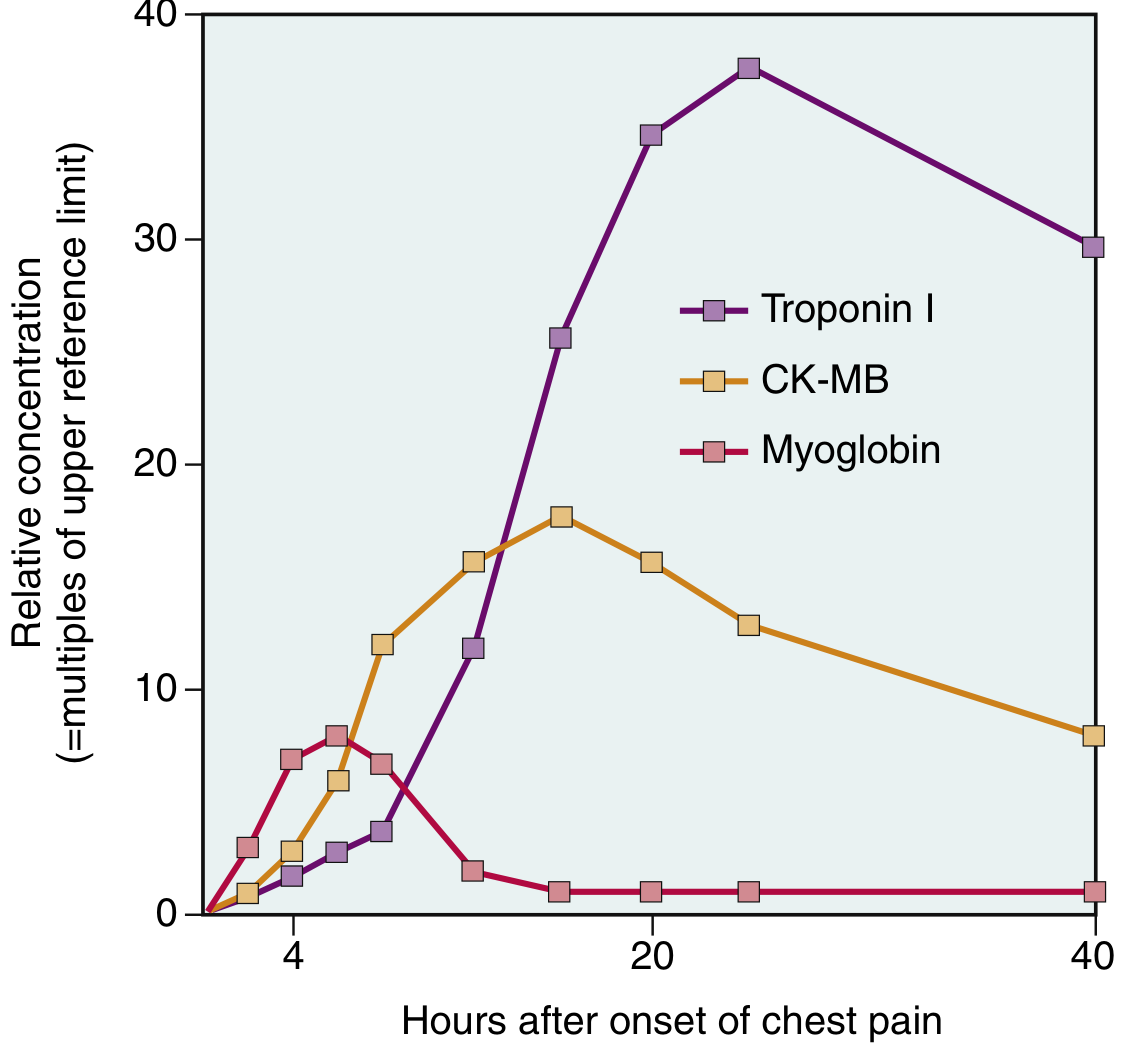
(Robbins & Kumar Basic Pathology, Fig. 9.13 - Acute increases in Troponin I, CK-MB, and Myoglobin following MI)
Enzyme/Biomarker Changes Summary
| Marker | Rise | Peak | Returns to Normal | Notes |
|---|---|---|---|---|
| Myoglobin | 1-4 hrs | 6-9 hrs | 24-36 hrs | Earliest to rise; low cardiac specificity |
| CK-MB (creatine kinase-MB isoform) | 2-4 hrs | 24-48 hrs | ~72 hrs | Traditionally used; now largely replaced by troponins |
| Troponin I (cTnI) | 2-4 hrs | ~48 hrs | 7-10 days | High cardiac specificity and sensitivity; preferred marker |
| Troponin T (cTnT) | 2-4 hrs | ~48 hrs | 7-14 days | High cardiac specificity; same kinetics as TnI |
| LDH (LDH-1 isoform) | 24-48 hrs | 3-5 days | 10-14 days | Useful for late diagnosis; LDH-1 > LDH-2 = "flipped LDH" |
| AST | 12-24 hrs | 36-48 hrs | 3-5 days | Non-specific; less used now |
Key points:
- TnI and TnT are normally absent from the circulation - any detectable level is significant
- Troponin is the gold-standard marker - persists 7-10 days, allowing late diagnosis
- With reperfusion: both troponin and CK-MB peak earlier due to rapid washout from necrotic tissue
- CK-MB returns to normal days earlier than LDH, making LDH useful for confirming an older infarct
Other Diagnostic Tools
- ECG: ST elevation (STEMI), ST depression/T-wave inversion (NSTEMI), new Q-waves (transmural necrosis), bundle branch block
- Echocardiography: Regional wall motion abnormalities (hypokinesis, akinesis, dyskinesis)
- Coronary angiography: Gold standard for identifying culprit artery; guides PCI
- Radionuclide scanning: Technetium-99m sestamibi perfusion imaging
- MRI: Most sensitive for detecting myocardial necrosis and viability
Complications of Myocardial Infarction
Nearly three-fourths of patients experience one or more complications. Three are potentially lethal:
Mechanical Complications (most dangerous)
| Complication | Timing | Details |
|---|---|---|
| Ventricular free wall rupture | 3-7 days (peak) | Least common but most fatal; causes hemopericardium and cardiac tamponade |
| Ventricular septal rupture (VSD) | 3-7 days | Most common rupture; harsh new systolic murmur |
| Papillary muscle rupture | 3-7 days | Causes acute severe mitral regurgitation; flash pulmonary edema |
Rupture timing: 3-7 days after MI when lysis of necrotic myocardium is maximal and the infarct has been converted to soft, friable granulation tissue (maximum vulnerability).
Functional Complications
- Contractile dysfunction / Cardiogenic shock: Proportional to extent of LV damage; cardiogenic shock occurs in ~10% of transmural MIs, typically when >40% of LV is damaged
- Papillary muscle dysfunction: Ischemia (without rupture) causes mitral regurgitation
- Right ventricular infarction: Isolated in 1-3% of MIs; right-sided failure, systemic hypotension
Arrhythmias
- Most common complication; major cause of early (pre-hospital) death
- Ventricular fibrillation: leading cause of death within first hour of MI
- Ventricular tachycardia, heart block, atrial fibrillation, sinus bradycardia (especially with inferior MIs via vagal stimulation)
Late / Chronic Complications
| Complication | Mechanism |
|---|---|
| Pericarditis (Dressler syndrome) | Autoimmune reaction 2-10 weeks post-MI; fibrinous epicardial inflammation |
| Mural thrombus | Wall motion abnormality over infarcted zone; risk of systemic embolism |
| Left ventricular aneurysm | Scar tissue bulges outward with each contraction; associated with persistent ST elevation |
| Progressive heart failure | Ventricular remodeling with myocyte hypertrophy and chamber dilation |
| Ventricular remodeling | Infarct expansion + reactive hypertrophy of non-infarcted segments |
Clinical Presentation (Brief)
- Classic: crushing/squeezing chest pain >30 minutes, radiating to left arm/jaw, diaphoresis, nausea/vomiting, dyspnea
- ~25% are entirely asymptomatic (especially diabetics with neuropathy) - discovered by ECG or biomarkers
- Posterior-inferior MI: nausea/vomiting predominant (vagal stimulation)
Sources: Robbins & Kumar Basic Pathology, pp. 358-360; Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 511-516
This is a shared conversation. Sign in to Orris to start your own chat.