I have my university exam of 1 st year of subject biochemistry These are the important question give me answer of this "Products of phenylalanine and tyrosine metabolism" and remeber it's a university examination The answer should be easy to memorize and follow the pattern of answer writing as in University paper
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
I now have all the information needed. Let me write the comprehensive university exam-style answer.
Products of Phenylalanine and Tyrosine Metabolism
Introduction
Phenylalanine and tyrosine are aromatic amino acids that share a common metabolic pathway. Phenylalanine is an essential amino acid (must be obtained from diet), while tyrosine is a non-essential amino acid synthesized from phenylalanine. Both are classified as glucogenic and ketogenic amino acids because their metabolism ultimately yields fumarate (glucogenic) and acetoacetate (ketogenic).
Part I: Conversion of Phenylalanine to Tyrosine
The first and most important step is the hydroxylation of phenylalanine to tyrosine:
- Enzyme: Phenylalanine hydroxylase (PAH) - a mixed-function oxidase
- Cofactor: Tetrahydrobiopterin (BH4), which is oxidized to dihydrobiopterin (BH2)
- BH4 regeneration: Dihydropteridine reductase (DHPR) regenerates BH4 from BH2 using NADPH
- Deficiency of PAH → Phenylketonuria (PKU)
Phenylalanine + O₂ + BH4 → Tyrosine + H₂O + BH2
The pathway diagram below shows the full degradation pathway:
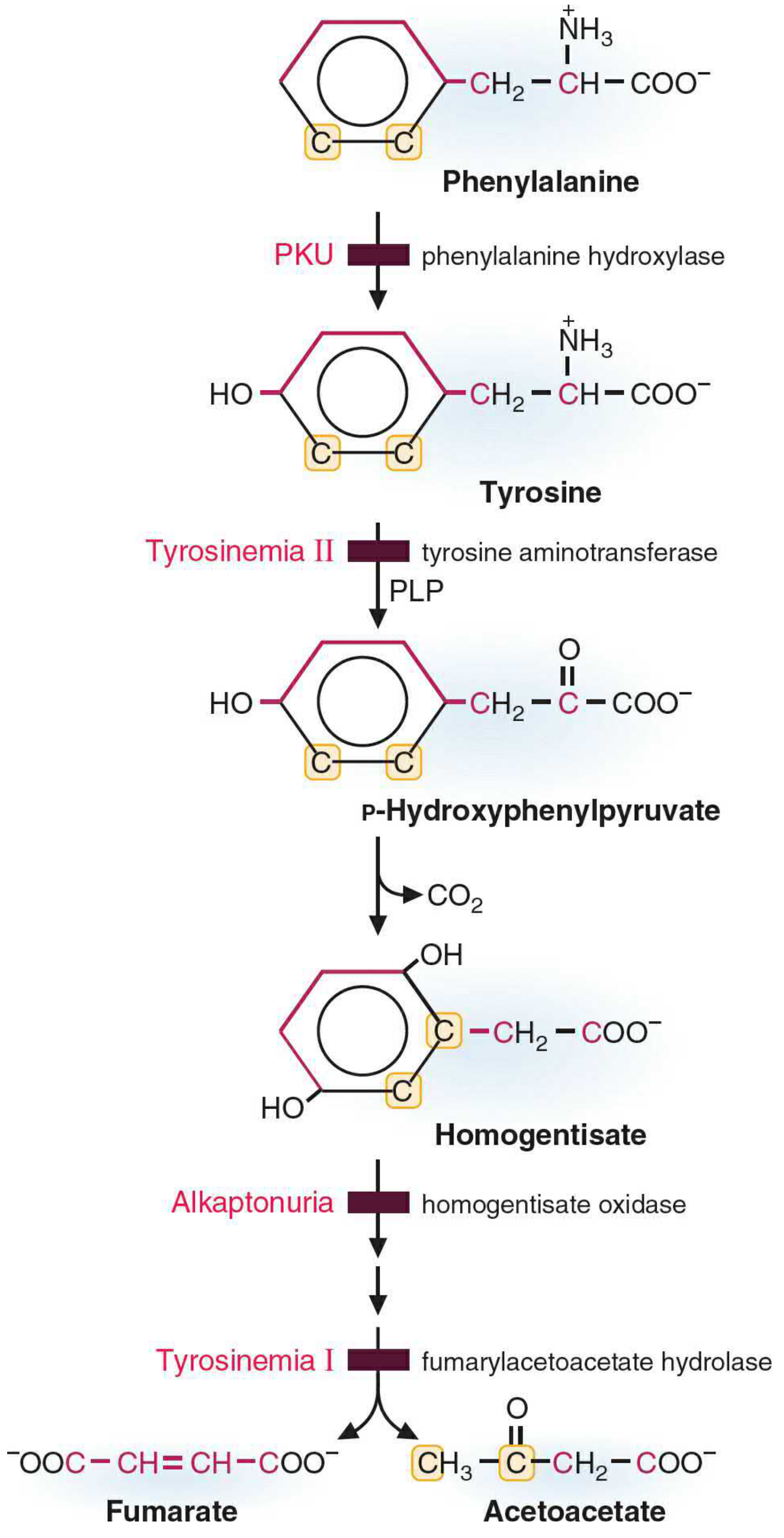
Part II: Products of Tyrosine Metabolism
Tyrosine serves as a precursor for four major categories of products:
A. Catabolic (Degradative) Products
The catabolic pathway of tyrosine proceeds as follows:
| Step | Reaction | Enzyme | Cofactor |
|---|---|---|---|
| 1 | Tyrosine → p-Hydroxyphenylpyruvate | Tyrosine aminotransferase (TAT) | PLP (Vit. B6) |
| 2 | p-Hydroxyphenylpyruvate → Homogentisate | p-Hydroxyphenylpyruvate dioxygenase | Vitamin C |
| 3 | Homogentisate → Maleylacetoacetate | Homogentisate oxidase | - |
| 4 | Maleylacetoacetate → Fumarylacetoacetate | Maleylacetoacetate isomerase | - |
| 5 | Fumarylacetoacetate → Fumarate + Acetoacetate | Fumarylacetoacetate hydrolase | - |
Final catabolic products:
- Fumarate - enters TCA cycle (glucogenic)
- Acetoacetate - a ketone body (ketogenic)
B. Catecholamines (Neurotransmitters and Hormones)
Tyrosine is the precursor for all catecholamines:
Tyrosine → L-DOPA → Dopamine → Norepinephrine → Epinephrine
- Tyrosine → L-DOPA: Tyrosine hydroxylase (requires BH4) - the rate-limiting step
- L-DOPA → Dopamine: DOPA decarboxylase (requires PLP/Vit. B6)
- Dopamine → Norepinephrine: Dopamine β-hydroxylase (requires Vitamin C)
- Norepinephrine → Epinephrine: PNMT (requires SAM as methyl donor)
Clinical note: L-DOPA (levodopa) is used in treatment of Parkinson's disease.
C. Melanin (Skin/Hair Pigment)
Tyrosine → DOPA → Dopaquinone → Melanin
- Enzyme: Tyrosinase (requires copper as cofactor)
- Deficiency of tyrosinase → Albinism (lack of skin, hair, and eye pigmentation)
D. Thyroid Hormones
Tyrosine residues in thyroglobulin are iodinated to form thyroid hormones:
- T3 (triiodothyronine) - more active form
- T4 (thyroxine) - main secreted form
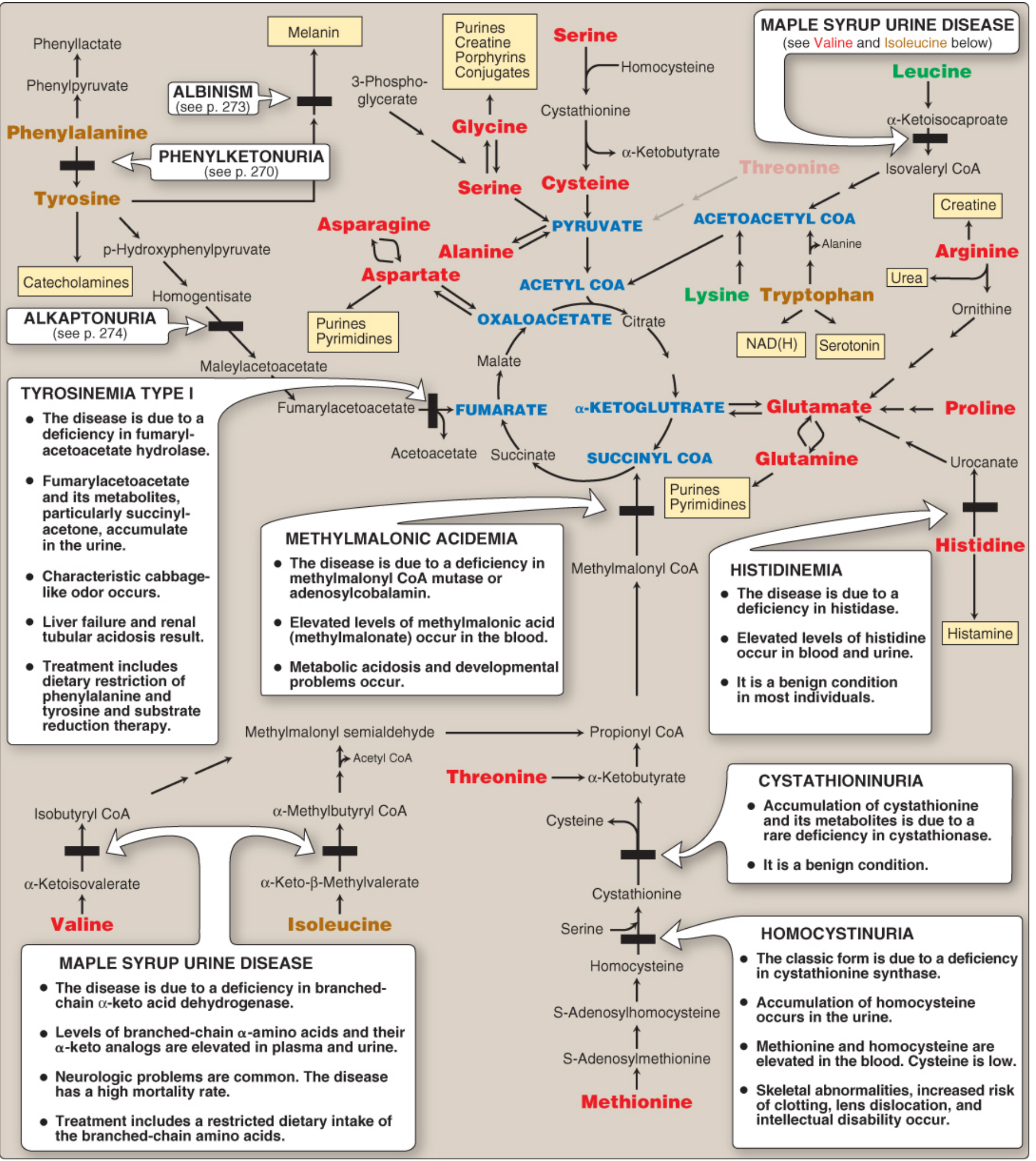
Part III: Inborn Errors of Metabolism
A very important exam topic - enzyme defects in this pathway:
| Disease | Deficient Enzyme | Accumulated Substance | Key Features |
|---|---|---|---|
| PKU (Phenylketonuria) | Phenylalanine hydroxylase (PAH) | Phenylalanine; phenylpyruvate, phenylacetate, phenyllactate in urine | Intellectual disability, musty odour of urine, fair skin/hair; treat with low-Phe diet + tyrosine supplementation |
| Alkaptonuria | Homogentisate oxidase | Homogentisate | Dark urine on standing (oxidizes to black pigment), ochronosis (darkening of cartilage), arthritis |
| Tyrosinemia Type I | Fumarylacetoacetate hydrolase | Fumarylacetoacetate + succinylacetone | Liver failure, renal tubular acidosis, characteristic cabbage-like odour; fatal in first year if untreated |
| Tyrosinemia Type II | Tyrosine aminotransferase | Tyrosine | Eye and skin lesions, neurological problems; treat with low-Tyr, low-Phe diet |
| Albinism | Tyrosinase | Tyrosine (in melanocytes) | Lack of melanin, photosensitivity, nystagmus |
Summary Diagram of All Products
The diagram below shows the full overview of products from phenylalanine and tyrosine metabolism including all associated diseases:
Quick Recall Table (For Exam)
| Product Category | Products |
|---|---|
| Catabolic end products | Fumarate + Acetoacetate |
| Neurotransmitters/Hormones | Dopamine, Norepinephrine, Epinephrine |
| Pigment | Melanin |
| Thyroid hormones | T3, T4 |
| Intermediate (disease marker) | Homogentisate (Alkaptonuria) |
Conclusion
Phenylalanine and tyrosine metabolism produces a wide variety of physiologically important products: the TCA cycle intermediates fumarate and acetoacetate (from catabolism), catecholamines (dopamine, norepinephrine, epinephrine), the pigment melanin, and thyroid hormones T3 and T4. Defects in specific enzymes of this pathway lead to clinically significant inborn errors including PKU, alkaptonuria, tyrosinemia, and albinism. Both amino acids are thus glucogenic and ketogenic, reflecting the dual metabolic fate of their carbon skeletons.
Sources: Basic Medical Biochemistry - A Clinical Approach, 6e | Lippincott Illustrated Reviews: Biochemistry, 8th ed
This is a shared conversation. Sign in to Orris to start your own chat.