A 4 year old boy presents to paediatrician for speech delay and behavioural difficulties
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Searching PubMed
speech delay child autism evaluation management
I now have a thorough set of information from multiple authoritative sources. Let me compile the comprehensive clinical response.
4-Year-Old Boy: Speech Delay + Behavioural Difficulties
This is a classic paediatric presentation requiring a systematic clinical approach. The combination of speech delay and behavioural difficulties at age 4 has a broad differential diagnosis, with Autism Spectrum Disorder (ASD) being the most important to evaluate - but hearing loss, intellectual disability, specific language impairment, and ADHD must all be considered.
Normal Language Milestones (for context)
| Age | Expected milestone |
|---|---|
| 3-4 months | Cooing (vowel sounds) |
| 6-12 months | Babbling; first words by ~12 months |
| 18 months | Word combinations; ~25% intelligible to strangers |
| 24 months | ~300 words; 50% intelligible |
| 30 months | ~500 words |
| 36 months | 75% intelligible to strangers |
| 4 years | Nearly 100% of words intelligible; most expressive language developed and appropriately used |
A 4-year-old who is not at this level has a clinically significant speech delay.
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Differential Diagnosis
1. Autism Spectrum Disorder (ASD) - TOP PRIORITY
The combination of speech/language delay + behavioural difficulties is the classic heralding presentation of ASD. While language impairment is not a DSM-5 core criterion, it is an extremely common associated feature.
DSM-5 Core Features (both domains required):
Domain 1 - Deficits in social communication:
- Impaired emotional reciprocity / social interaction
- Reduced or absent nonverbal communication (eye contact, gestures, facial expression)
- Difficulty forming and maintaining relationships
Domain 2 - Restricted, repetitive behaviours/interests:
- Stereotyped motor movements
- Rigid adherence to routines/rituals
- Restricted, fixed interests
- Hyper- or hyporeactivity to sensory input
Key points for this 4-year-old:
-
~1/3 of ASD children have co-occurring intellectual disability
-
Up to 25% show language regression (words present then lost)
-
"Theory of mind" deficit: difficulty inferring others' feelings leads to social awkwardness
-
Exploratory play is restricted; toys used in ritualistic, idiosyncratic ways
-
Symptoms begin in the early developmental period but may not be clearly recognized until school age in milder cases
-
Kaplan and Sadock's Synopsis of Psychiatry
2. Isolated / Benign Developmental Speech Delay
-
A subset of otherwise neurologically normal children who are "late talkers"
-
3:1 male predominance, often positive family history
-
Comprehension and general intelligence are normal - this is the key differentiator
-
Communication by gestures is facile
-
When speech eventually develops, it may progress rapidly
-
However, many such children later have dyslexia/dysgraphia, so follow-up is essential
-
(Notably, Albert Einstein reportedly did not speak until age 4)
-
Adams and Victor's Principles of Neurology
3. Hearing Loss / Congenital Deafness - MUST EXCLUDE FIRST
-
Estimated 3 million American children have hearing defects; 0.1% of school-age children are deaf
-
Risk factors: familial deaf mutism, congenital rubella, erythroblastosis fetalis, chronic bilateral ear infections (very common), meningitis history, ototoxic drug exposure
-
The deaf child typically: appears eager to communicate via gesture/facial expression, has normal social milestones, has normal intelligence (Leiter scale)
-
Babbling is normal up to 6 months then becomes stereotyped and restricted
-
Audiometry (and BAER if needed) is mandatory in any child with speech delay
-
Adams and Victor's Principles of Neurology
4. Global Developmental Delay / Intellectual Disability
- 35-50% of children with pathological speech delay have global developmental delay or cerebral palsy
- Developmental milestones across multiple domains (motor, cognitive, social) are all delayed
- Unlike isolated speech delay, general intelligence is also impaired
5. Specific Language Impairment (SLI) / Communication Disorder
- Selective delay in language acquisition with relatively preserved nonverbal intelligence
- No social communication deficits (distinguishes from ASD)
- No hearing loss
6. Other considerations:
- ADHD (behavioural difficulties, impaired attention affecting language acquisition)
- Selective mutism (speaks in some settings but not others)
- Childhood disintegrative disorder (normal development x2 years, then regression)
- Fragile X syndrome (most common inherited intellectual disability; characteristic physical features)
- Landau-Kleffner syndrome (acquired epileptic aphasia with bitemporal EEG discharges)
- Lead toxicity, hypothyroidism, iron deficiency (metabolic/toxic causes)
History - Key Questions
Developmental history:
- Age of first words? First sentences?
- Regression? (Loss of words previously acquired - red flag for ASD)
- Motor milestones (walking, fine motor)?
- Bladder/bowel control?
Speech/language characterization:
- Does he attempt to communicate? How (words, gestures, pointing)?
- Does he understand commands? (receptive vs expressive language)
- Is speech intelligible? Echolalia present?
Behavioural profile:
- Specific behavioural difficulties (tantrums, aggression, rigidity, inattention)?
- Response to name when called?
- Eye contact?
- Repetitive movements (hand flapping, rocking)?
- Restricted interests? Insistence on sameness/routines?
- Play: imaginative/symbolic play present?
- Social interest in peers and adults?
Hearing:
- Passes hearing at birth (newborn screen)?
- Parents' concern about hearing? Turns to sounds?
- Recurrent ear infections/glue ear?
Medical/family history:
- Perinatal complications, prematurity?
- Seizures?
- Family history of ASD, speech delay, intellectual disability, learning disorders?
- Consanguinity?
Examination
General: dysmorphic features (Fragile X, Down syndrome, Angelman syndrome), head circumference (macrocephaly in Fragile X, microcephaly)
Neurology: tone, reflexes, coordination, focal signs
Hearing: response to voice/sound at exam, otoscopy (otitis media with effusion)
Behavioural observation during consultation:
- Eye contact, joint attention (pointing to share interest)
- Response to name
- Play quality (functional vs repetitive)
- Social reciprocity with parent and examiner
Skin: ash-leaf macules (tuberous sclerosis)
Investigations
| Investigation | Rationale |
|---|---|
| Formal audiological assessment (audiometry/BAER) | Exclude hearing loss - mandatory |
| Developmental screening tool (M-CHAT-R, Ages & Stages Questionnaire) | Structured ASD/developmental screen |
| Formal ASD assessment (ADOS-G - Autism Diagnostic Observation Schedule, ADI-R) | Gold standard diagnostic tools |
| Developmental/IQ testing | Quantify cognitive function |
| Chromosomal microarray / karyotype | Fragile X, chromosomal syndromes |
| FMR1 gene analysis | Fragile X (most common if dysmorphic) |
| Thyroid function tests | Hypothyroidism causing developmental delay |
| Lead level | If risk factors present |
| EEG | If seizures suspected / Landau-Kleffner |
| MRI brain | If focal neurological signs, regression, or dysmorphic features |
| Metabolic screen (urine amino acids, organic acids) | If clinical features suggest IEM |
Management
Immediate referrals (do not wait for diagnosis to refer):
- Audiology - hearing assessment urgently
- Speech and Language Therapy (SLT) - both diagnostic and therapeutic
- Developmental Paediatrics / Child Psychiatry - formal ASD/developmental evaluation
- Early intervention programme - services available without waiting for a formal diagnosis in most health systems
If ASD confirmed:
Behavioural/Educational Interventions (most evidence-based):
- Applied Behaviour Analysis (ABA) - structured operant conditioning; evidence for improving adaptive behaviours
- Early Start Denver Model (ESDM) - for very young children; naturalistic settings (home, daycare); parents as co-therapists; integrates ABA with play
- Pivotal Response Training (PRT) - parent-delivered; targets gateway social behaviours
- Social Skills Training - group-based; teaches initiation, greetings, emotion recognition
- Cognitive-Behavioural Therapy (CBT) - for co-morbid anxiety, OCD, repetitive behaviours
Pharmacological (symptom-specific, no medication treats core ASD):
- Risperidone or aripiprazole: for significant irritability, aggression, self-injurious behaviour
- Melatonin: for insomnia (very prevalent in ASD)
- Stimulants/atomoxetine: for co-morbid ADHD symptoms
Family support:
- Parent training (Hanen "More Than Words" programme)
- Carer psychological support
- Educational placement in specialist or supported mainstream setting
If isolated speech delay:
- SLT (direct and/or parent-mediated)
- Monitor for learning difficulties (dyslexia, dysgraphia) at school age
- Hearing aids if any hearing deficit
If hearing loss:
- ENT referral (grommets if otitis media with effusion)
- Hearing aid fitting
- Specialist hearing/speech education programme
Red Flags Warranting Urgent Assessment
- No babbling by 12 months
- No single words by 16 months
- No two-word phrases by 24 months
- Any loss of previously acquired language or social skills at any age
- No response to name
- Absence of joint attention (pointing, showing)
- Regression of any developmental skill
Sources:
- Kaplan and Sadock's Synopsis of Psychiatry (Section 2.3 - Autism Spectrum Disorder; Communication Disorders)
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry (Language Development)
- Adams and Victor's Principles of Neurology, 12th Edition (Developmental Speech Delay; Congenital Deafness)
Medical record reveals increasing head circumference and recurrent respiratory infections
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
I now have comprehensive information to give a full, focused answer. Let me compile the complete clinical response.
Updated Assessment: Macrocephaly + Recurrent Respiratory Infections Narrows the Diagnosis Significantly
The addition of progressive macrocephaly and recurrent respiratory infections to speech delay and behavioural difficulties in a 4-year-old boy is a classic syndromic constellation pointing strongly toward a Mucopolysaccharidosis (MPS) - a lysosomal storage disorder caused by defective degradation and progressive accumulation of glycosaminoglycans (GAGs).
The male sex specifically raises Hunter Syndrome (MPS II) as the leading diagnosis, as it is the only X-linked MPS - all others are autosomal recessive.
The Pathophysiology
MPS disorders arise from deficiency of a specific lysosomal enzyme needed to break down sulfated polysaccharides (GAGs: dermatan sulfate, heparan sulfate, keratan sulfate, chondroitin sulfate). The undegraded GAGs accumulate progressively in lysosomes throughout the body - connective tissue, skeleton, airways, liver, spleen, heart, and CNS - producing a chronic, multisystem, progressive disease.
- Goldman-Cecil Medicine
Differential Diagnosis: The MPS Spectrum
| MPS Type | Eponym | Enzyme Deficient | GAG Excreted | CNS | Corneal Clouding | Key Distinguisher |
|---|---|---|---|---|---|---|
| I | Hurler | α-L-iduronidase | Dermatan + heparan sulfate | Severe | YES | Most severe; death 1st decade |
| I S | Scheie | α-L-iduronidase | Dermatan + heparan sulfate | Normal intelligence | Yes | Mild form; normal IQ |
| II | Hunter | Iduronate-2-sulfatase | Dermatan + heparan sulfate | Variable | NO | X-linked; males only |
| III | Sanfilippo | 4 possible enzymes | Heparan sulfate | Severe | Absent | Behaviour/CNS >>> somatic features |
| IV | Morquio | N-acetylgalactosamine-6-sulfatase | Keratan sulfate | Normal | No | Severe skeletal dysplasia |
| VI | Maroteaux-Lamy | Arylsulfatase B | Dermatan sulfate | Normal | Yes | Somatic features, normal IQ |
- Adams and Victor's Principles of Neurology; Cummings Otolaryngology; Emery's Elements of Medical Genetics
The Two Leading Diagnoses in This Boy
1. Hunter Syndrome (MPS II) - MOST LIKELY
Inheritance: X-linked recessive - affects males exclusively in classic form
Enzyme deficiency: Iduronate-2-sulfatase (IDS gene mutation)
GAGs accumulated: Dermatan sulfate + heparan sulfate (excreted in urine)
Presentation (ages 2-5 years):
- Recurrent infections - upper respiratory, otitis media (due to GAG accumulation in upper airway mucosa and eustachian tube dysfunction)
- Increasing head circumference (macrocephaly from cranial vault expansion + communicating hydrocephalus)
- Hearing loss - mixed conductive and sensorineural
- Coarsening of facial features (full lips, broad nose, wide-set eyes, large tongue)
- Hepatosplenomegaly
- Joint stiffness (claw hand, limited range of motion)
- Short stature / poor growth
- Speech delay + behavioural difficulties (hyperactivity, aggression)
- Progressive mental deterioration in severe form
Hunter vs Hurler: Hunter is clinically similar to Hurler but:
- No corneal clouding (key differentiating feature)
- Slightly milder course
- Survival to adulthood possible in attenuated form
Skin finding (pathognomonic ~10%): Ivory-white/flesh-coloured pebbly papules and nodules in a cobblestone/reticular pattern on upper back, neck, chest, proximal arms - histologically showing dermal mucin accumulation
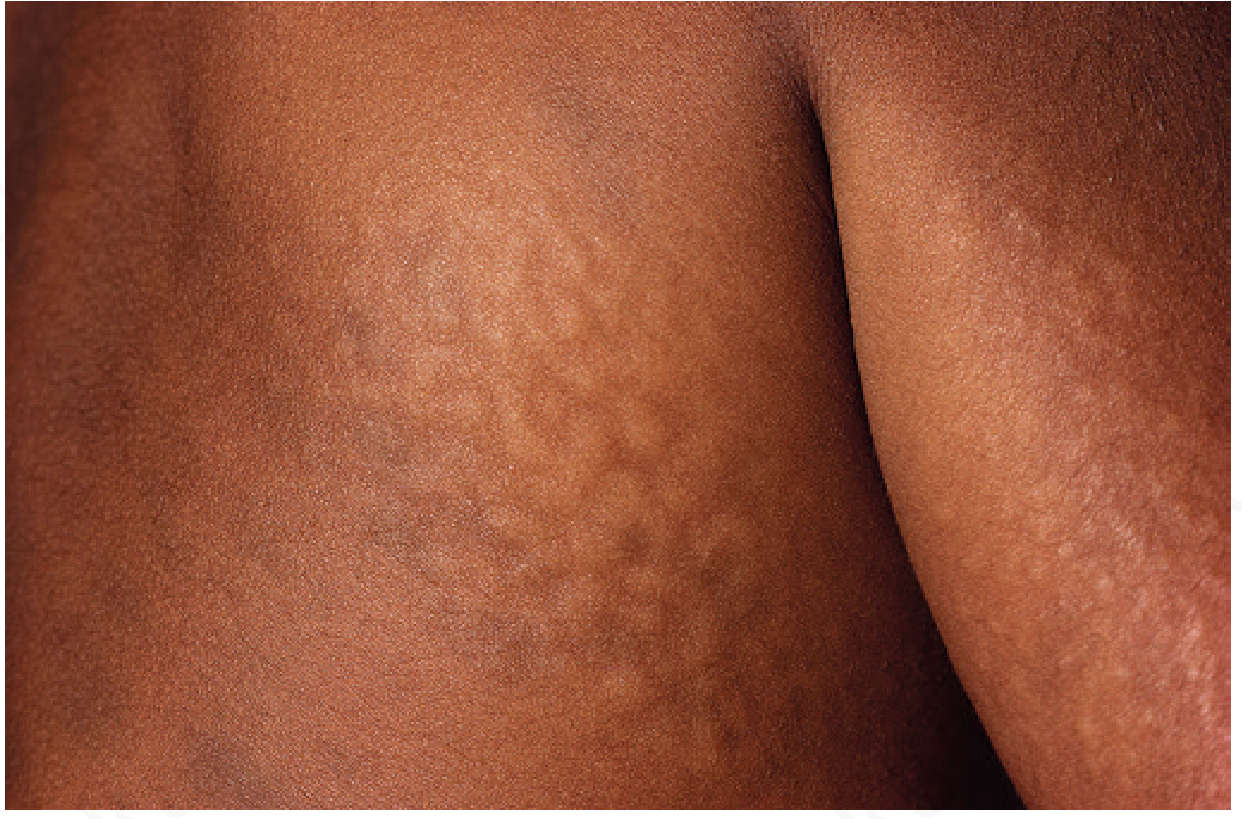
Two clinical forms:
-
Severe (neuronopathic): Progressive cognitive decline, death mid-teens
-
Attenuated: Relatively preserved intelligence, survival into middle age
-
Adams and Victor's Principles of Neurology; Emery's Elements of Medical Genetics
2. Sanfilippo Syndrome (MPS III) - IMPORTANT DIFFERENTIAL
- Most common MPS overall
- Presents age 2-3 years with progressive intellectual deterioration
- Behavioural problems are prominent and often severe (hyperactivity, aggression, sleep disturbance - sometimes the presenting complaint)
- Somatic features are milder - subtle coarsening, mild hepatosplenomegaly; no corneal clouding
- Heparan sulfate excreted in urine
- 4 subtypes (A, B, C, D) by enzyme defect - all phenotypically similar
- Progressive deterioration into vegetative state, death in third decade
- No effective enzyme replacement therapy currently available
3. Hurler Syndrome (MPS I) - Less likely given male sex and absence of corneal clouding yet
- Autosomal recessive - affects both sexes
- Most severe MPS; begins in first year of life
- Corneal clouding is characteristic (differentiates from Hunter)
- Gargoyle facies, macrocephaly, kyphosis, broad stubby hands
- Conductive deafness, corticospinal signs
- Hepatosplenomegaly, valvular heart disease
- Death first decade
Additional Features to Elicit Now
On history:
- Family history of affected males (maternal uncles/cousins) - strongly suggests Hunter (X-linked)
- Age of symptom onset and progression (is it worsening?)
- Snoring, obstructive sleep apnoea (from upper airway GAG deposition)
- Diarrhoea (common in Hunter)
- Joint stiffness, difficulty walking
- Skin changes (pebbly papules)
On examination:
- Head circumference - plot on centile chart, confirm progressive macrocephaly
- Facial features: coarsening (flat nasal bridge, full lips, prominent brow, large tongue)
- Corneas: slit-lamp examination - clouding = Hurler; clear = Hunter
- Hepatosplenomegaly - palpate abdomen
- Joints: range of motion, claw hand, genu valgum
- Cardiovascular: heart murmur (valvular disease)
- Spine: kyphosis/gibbus deformity
- Hearing: examine ears, formal audiology
- Skin: look for pebbly papules (Hunter)
- Airway: tonsillar/adenoidal hypertrophy
Investigations
| Investigation | Finding Expected | Rationale |
|---|---|---|
| Urine GAG spot test (urinary glycosaminoglycan quantification + fractionation) | Elevated dermatan + heparan sulfate | First-line screening |
| Leukocyte enzyme assay - iduronate-2-sulfatase | Absent/markedly reduced | Confirms Hunter syndrome |
| IDS gene mutation analysis | Pathogenic variant | Confirms Hunter, identifies carrier females |
| Leukocyte α-L-iduronidase assay | Reduced - if Hurler suspected | Differential |
| Skeletal survey (dysostosis multiplex) | Broad ribs ("oar-shaped"), J-shaped sella, vertebral beaking, shortened metacarpals | Radiological pattern of MPS |
| MRI brain | Communicating hydrocephalus, white matter changes, perivascular spaces | Assess CNS involvement |
| Audiological assessment | Mixed conductive + sensorineural hearing loss | Common in MPS |
| Echocardiogram | Valvular thickening/disease, cardiomyopathy | Cardiovascular involvement |
| Liver function + USS abdomen | Hepatosplenomegaly | Confirm organomegaly |
| Ophthalmology (slit-lamp) | Corneal clouding (Hurler) vs clear (Hunter) | Critical differentiator |
| Chromosomes / CGH array | Rule out chromosomal causes | If diagnosis uncertain |
| Newborn screening (if available) | - | Some US programs screen for MPS I |
Management
Disease-Modifying Therapy
1. Enzyme Replacement Therapy (ERT)
- Hunter (MPS II): Idursulfase (Elaprase) - weekly IV infusion; may delay somatic features, but limited CNS penetration (does not cross blood-brain barrier effectively)
- Hurler (MPS I): Laronidase (Aldurazyme) - recombinant α-L-iduronidase
- ERT is available for MPS I, II, IVA, VI, VII
- Critical: must start before significant neurological decline for maximum benefit
- Neither ERT nor bone marrow transplantation has been shown effective in Sanfilippo (MPS III)
2. Haematopoietic Stem Cell Transplantation (HSCT)
- Hurler (MPS I): HSCT (cord blood from unrelated donors) is the treatment of choice when performed early (before age 2-2.5 years, before significant CNS decline)
- Less effective for Hunter or Sanfilippo
- Does not improve eye or bone disease in Hurler
3. Gene Therapy
-
Experimental; promising for MPS I and MPS VI
-
Several clinical trials ongoing
-
Goldman-Cecil Medicine; Adams and Victor's Principles of Neurology
Supportive/Symptomatic Management
| Domain | Intervention |
|---|---|
| Airway | ENT review for tonsillectomy/adenoidectomy; CPAP for obstructive sleep apnoea; high anaesthetic risk - avoid if possible, use experienced team |
| Hearing | Grommets for otitis media with effusion; hearing aids |
| Cervical myelopathy | Monitor MRI cervical spine (MPS I, II, VI); surgical decompression if needed |
| Cardiac | Echocardiography, cardiology input for valvular disease |
| Orthopaedic | Physiotherapy, joint management, hip/spine surgery as needed |
| Neurodevelopmental | Speech therapy, special education, behavioural support |
| Carpal tunnel | Surgical release (common in adults with MPS) |
| Genetic counselling | Family counselling; carrier testing for maternal relatives (Hunter is X-linked - maternal aunts, female cousins at risk) |
Anaesthetic alert: All MPS patients are high anaesthetic risk due to difficult airway (macroglossia, short neck, tracheal narrowing), atlantoaxial instability, and cardiac involvement. This must be flagged prominently in the medical record.
Summary: The Diagnostic Hierarchy in This Boy
4-year-old boy
├── Speech delay + behavioural difficulties
├── PROGRESSIVE MACROCEPHALY → Storage / infiltrative / CSF disorder
└── RECURRENT RESPIRATORY INFECTIONS → Airway GAG deposition
Combined pattern → MUCOPOLYSACCHARIDOSIS
Male sex + X-linked pattern → HUNTER SYNDROME (MPS II) most likely
Confirm with: Urine GAGs → Leukocyte iduronate-2-sulfatase → IDS gene analysis
If corneal clouding present → Hurler (MPS I)
If somatic features minimal, behaviour dominates → Sanfilippo (MPS III)
Sources:
- Adams and Victor's Principles of Neurology, 12th ed. (MPS II/III sections)
- Goldman-Cecil Medicine (Mucopolysaccharidoses, Lysosomal Storage Disorders)
- Emery's Elements of Medical Genetics and Genomics (Hunter, Hurler, Sanfilippo)
- Cummings Otolaryngology Head and Neck Surgery (MPS - otologic manifestations)
- Andrews' Diseases of the Skin (Hunter syndrome skin findings)
6 year old boy is brought in by his parents due to concerns about developmental delays and behavioural difficulties in schools. He has history of speech and language delays difficulties with social interaction hyperactivity and repetitive behaviours . His mother mentions her brother had similar learning difficulties and her father developed tremors and balance problems in his late sixties. On exam he is noted to have long narrow face with prominent ears and prominent jaw
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Loading Image
Loading Image
I now have all the information needed for a complete, authoritative response.
Diagnosis: Fragile X Syndrome (FXS)
This presentation is clinically diagnostic of Fragile X Syndrome - the most common inherited cause of intellectual disability and the most common single-gene cause of ASD. Every feature of this case locks in:
| Clinical Feature | Significance |
|---|---|
| 6-year-old male | FXS affects males predominantly (X-linked) |
| Long narrow face, prominent ears, prominent jaw | Classic FXS facial dysmorphism |
| Speech/language delay + social difficulties + hyperactivity + repetitive behaviours | Core neurodevelopmental phenotype |
| Maternal uncle with learning difficulties | X-linked inheritance - maternal uncle = mother's brother |
| Maternal grandfather with tremor + balance problems in late 60s | FXTAS (Fragile X-associated Tremor/Ataxia Syndrome) - premutation in maternal grandfather |
This three-generation pedigree (grandfather with FXTAS → carrier mother → affected grandson) is textbook Fragile X.
Genetics and Molecular Basis
The FMR1 Gene
- Locus: Xq27.3
- Gene: FMR1 (Fragile X Messenger Ribonucleoprotein 1)
- Mutation type: CGG trinucleotide repeat expansion in the 5' untranslated region
- Inheritance: X-linked (with unusual features - see below)
CGG Repeat Thresholds
| Category | CGG Repeat Number | Clinical Consequence |
|---|---|---|
| Normal | 6-55 | No disease |
| Intermediate / "grey zone" | 45-54 | Unstable, may expand |
| Premutation | 55-200 | Carrier; FXTAS (males) / FXPOI (females) |
| Full mutation | >200 (up to 4000) | Fragile X Syndrome |
- Robbins, Cotran & Kumar Pathologic Basis of Disease; Emery's Elements of Medical Genetics
How the Full Mutation Causes Disease
When CGG repeats exceed ~230, hypermethylation of the promoter region silences the FMR1 gene entirely → loss of FMRP protein.
FMRP (Fragile X Mental Retardation Protein) is normally:
- Most abundant in brain and testes (explaining the two organs most affected)
- A cytoplasmic RNA-binding protein that transports mRNAs to neuronal dendrites
- A translation regulator at synapses - it suppresses protein synthesis via group I metabotropic glutamate receptor (mGluR) signalling
- Essential for synaptic plasticity - the molecular basis of learning and memory
Without FMRP: Over-translation of synaptic proteins → loss of synaptic plasticity → intellectual disability
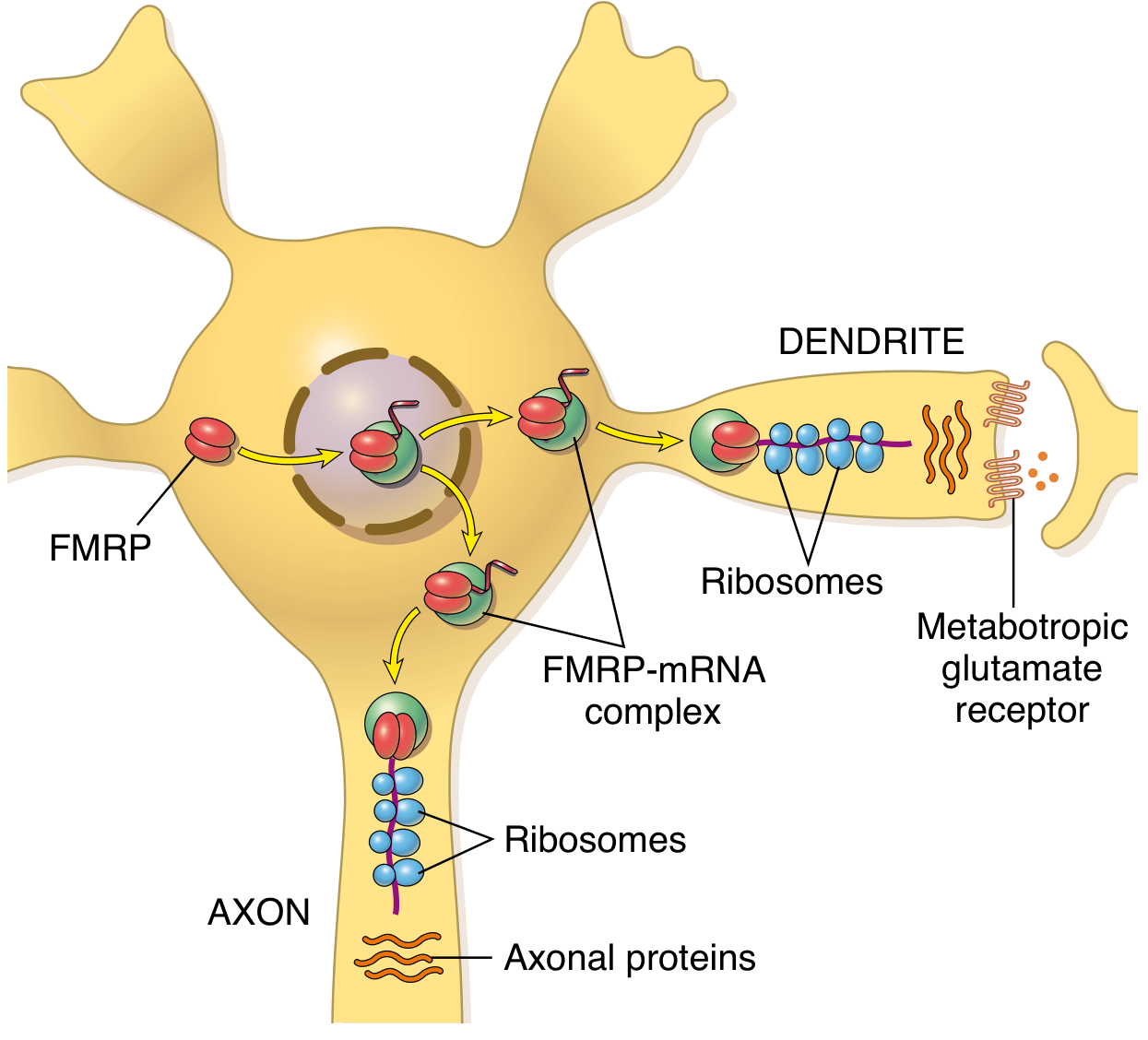
The Unusual Inheritance Pattern
FXS does not follow simple X-linked recessive rules. Three peculiarities explain this pedigree:
1. Anticipation
Expansion of CGG repeats worsens with each successive generation. The key rule: premutation expands to full mutation ONLY during female meiosis (oogenesis) - essentially never during male meiosis (spermatogenesis).
2. The Grandfather's Role (Premutation = FXTAS)
- The maternal grandfather has 55-200 CGG repeats (premutation)
- His premutation was passed to his daughter (the mother) unchanged - she is a carrier with premutation
- During the mother's oogenesis, the premutation expanded dramatically to a full mutation (>200 repeats)
- Her son inherited the full mutation → Fragile X Syndrome
- The grandfather himself never had FXS but developed FXTAS (toxic gain-of-function of the abnormal FMR1 mRNA - see below)
3. Normal Transmitting Males
Carrier males with the premutation transmit it to all daughters unchanged but have normal intelligence. These daughters then risk having affected sons if the premutation expands during their oogenesis. The risk of expansion from premutation to full mutation increases with increasing premutation size.
4. Penetrance in Females
- 30-50% of females who inherit the full mutation have intellectual disability (compared with nearly 100% of males) - due to X-inactivation of the unaffected X in some cells providing partial protection
The Three FMR1-Associated Disorders
The FMR1 locus causes three clinically distinct conditions depending on repeat size and mechanism:
CGG Repeat Size Person Disorder
────────────────────────────────────────────────────────
>200 (full) Males FRAGILE X SYNDROME (FXS) - loss of FMRP
>200 (full) Females Mild-moderate ID in ~40-50%
55-200 (pre) Males >50 yrs FXTAS - toxic gain-of-function
55-200 (pre) Females FXPOI (Primary Ovarian Insufficiency) ~20%
FXTAS - Explaining the Grandfather
Fragile X-associated Tremor/Ataxia Syndrome (FXTAS)
-
Affects ~50% of male premutation carriers; onset after age 50 (peaks in 60s-70s)
-
Mechanism: Toxic gain-of-function - the expanded FMR1 mRNA is NOT silenced (unlike full mutation) but is over-transcribed; the abnormal CGG-containing mRNA sequesters RNA-binding proteins into intranuclear inclusions in neurons - toxic neurodegeneration
-
Clinical features:
- Intention/kinetic tremor (this grandfather's presenting complaint)
- Gait ataxia / balance problems (this grandfather's second complaint)
- Executive cognitive dysfunction
- Parkinsonism
- Peripheral neuropathy
- Dysautonomia, erectile dysfunction
- Bilateral cerebellar/white matter hyperintensities on T2 MRI
-
Bradley and Daroff's Neurology; Robbins & Kumar
FXPOI - The Mother's Risk
The mother (carrier with premutation) has approximately:
- 20% risk of Fragile X-associated Primary Ovarian Insufficiency (FXPOI) - premature ovarian failure before age 40 (vs ~1% in general population)
- Elevated FSH, decreased anti-Müllerian hormone, early menopause (~5 years earlier than average)
- This should be discussed with the mother as part of genetic counselling
Full Clinical Features of FXS in This Boy
Facial Dysmorphism (classic triad confirmed on exam):
- Long narrow face
- Prominent ears (large, may be low-set)
- Prominent jaw (prognathism)
- Also: high forehead, slightly large head
Post-pubertal feature (to watch for):
- Macroorchidism (large testes) - most reliable physical sign in adult males; due to FMRP deficiency in testicular tissue
Connective tissue features:
- Hyperextensible joints
- Skin striae
- Mitral valve prolapse
Neurodevelopmental profile:
-
Intellectual disability - moderate to severe in males (IQ typically 40-70)
-
Speech delay - halting, repetitive, perseverative speech quality
-
Autism spectrum features - 50-75% of FXS males; eye gaze avoidance, poor social reciprocity
-
ADHD/hyperactivity - very common (90% show behavioural difficulties)
-
Anxiety - prominent, especially social anxiety
-
Sensory hypersensitivity - tactile, auditory
-
Epilepsy - ~30% of cases
-
Emery's Elements of Medical Genetics; Robbins & Kumar
Investigations
| Test | Purpose | Method |
|---|---|---|
| FMR1 CGG repeat analysis by PCR | First-line: detects normal and premutation alleles | Rapid, quantifies repeat number |
| Southern blotting | Detects full mutations (>200 repeats) and methylation status; PCR often fails to amplify very large expansions | Definitive for full mutation |
| Both PCR + Southern blot | Gold standard combination for complete characterisation | |
| Chromosomal microarray | Rule out co-existing chromosomal abnormality | If features atypical |
| EEG | If seizures suspected (~30% incidence) | |
| Echocardiogram | Mitral valve prolapse | |
| Developmental / IQ assessment | Quantify cognitive profile | |
| Audiology | Hearing assessment | |
| FMR1 testing of mother | Confirm carrier status; determine premutation vs full mutation | Essential for genetic counselling |
| FMR1 testing of grandfather | Confirm FXTAS diagnosis; establish premutation |
Note on cytogenetics: The historical "fragile site" on karyotype under folate-deficient culture conditions was the original detection method. This is now superseded by DNA-based FMR1 repeat analysis, which is more sensitive and specific.
Management
FXS has no cure currently - management is symptomatic and supportive, targeting each domain:
Neurodevelopmental
| Domain | Intervention |
|---|---|
| Speech and language | Speech and language therapy - early, intensive |
| Cognitive/learning | Special education support; individualised education plan (IEP) |
| Occupational therapy | Sensory integration therapy; fine motor skills |
| Behavioural | ABA (Applied Behaviour Analysis); structured routines |
Pharmacological (symptom-targeted)
| Symptom | Drug |
|---|---|
| Hyperactivity/ADHD | Methylphenidate or amphetamines (used cautiously - may worsen anxiety in FXS) |
| Anxiety | SSRIs (sertraline, fluoxetine) - evidence for anxiety and repetitive behaviours |
| Aggression/irritability | Risperidone or aripiprazole (atypical antipsychotics) |
| Seizures | Anticonvulsants (valproate, carbamazepine) |
| Sleep disturbance | Melatonin |
Investigational / Targeted Therapies
- mGluR5 antagonists (e.g. mavoglurant, arbaclofen): Rationale from the mGluR theory - blocking excessive glutamate signalling should restore synaptic balance. Trials have been disappointing in adults but ongoing in children
- GABA-B agonists (arbaclofen): Addresses GABA-glutamate imbalance in FXS
- Gene therapy / CRISPR: Preclinical research to restore FMRP expression
For the Family
- Genetic counselling: All family members at risk should be offered FMR1 testing
- The mother (likely premutation carrier - confirm with testing)
- Maternal aunts (may carry premutation - ovarian insufficiency risk)
- Maternal grandfather (premutation confirmed by FXTAS)
- Pre-implantation genetic diagnosis (PGD) / prenatal diagnosis available for future pregnancies
- FXTAS counselling for grandfather - refer to neurology; no disease-modifying treatment but supportive care (propranolol for tremor, physiotherapy for balance)
- FXPOI counselling for mother - endocrine monitoring, fertility planning, HRT consideration
The Pedigree in This Case - Explained
GENERATION I:
Grandfather ─────── Grandmother
(premutation: FXTAS (normal)
tremor + ataxia, 60s)
│
┌──────┴──────┐
│ │
GENERATION II:
Mother (carrier Uncle (affected: learning
premutation → full difficulties - inherited
mutation during her premutation from grandfather,
oogenesis) not fully expanded as
transmitted paternally*)
│
GENERATION III:
THIS BOY (full mutation, FXS)
Note: The maternal uncle's learning difficulties may reflect a premutation effect or a separate full mutation passed via the mother - the uncle is the son of the grandmother, and his X came from the grandfather (premutation). Males transmit the premutation with minimal change, so the uncle received a premutation - premutation in males does not usually cause intellectual disability but can cause subtle cognitive effects and FXTAS risk later.
Key Concepts Summary
| Feature | Detail |
|---|---|
| Gene | FMR1 (Xq27.3) |
| Mutation | CGG trinucleotide repeat expansion |
| Full mutation | >200 repeats → promoter methylation → FMR1 silencing → no FMRP |
| Premutation | 55-200 repeats → toxic mRNA gain-of-function → FXTAS / FXPOI |
| Protein lost | FMRP - synaptic mRNA transport and translation regulator |
| Mechanism of ID | Loss of synaptic plasticity via mGluR pathway dysregulation |
| Frequency | 1 in 3000-4000 male births; 1 in 5000 overall |
| Most common | Most common inherited cause of ID; most common single-gene cause of ASD |
| Diagnosis | PCR + Southern blot of FMR1 CGG repeat number + methylation status |
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (FXS molecular mechanisms)
- Thompson & Thompson Genetics and Genomics in Medicine, 9th ed.
- Emery's Elements of Medical Genetics and Genomics (clinical features, FXTAS)
- Bradley and Daroff's Neurology in Clinical Practice (FXTAS)
- Henry's Clinical Diagnosis and Management by Laboratory Methods (laboratory diagnosis)
This is a shared conversation. Sign in to Orris to start your own chat.