what is retinal pigmentosa
Reading File
Finding Sources
Searching PubMed
"retinitis pigmentosa"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information to provide a thorough answer. Note: the user wrote "retinal pigmentosa" which is a common misspelling - the correct term is retinitis pigmentosa (RP).
Retinitis Pigmentosa (RP)
Note: You likely meant retinitis pigmentosa - "retinal pigmentosa" is not a standard medical term. The name itself is a misnomer, since RP is not an inflammatory disease ("retinitis" implies inflammation, which is incorrect).
What Is It?
Retinitis pigmentosa is a broad term for a clinically and genetically diverse group of inherited diffuse retinal degenerations that primarily affect the rod photoreceptors first, followed by progressive degeneration of cones and the retinal pigment epithelium (RPE). It is the most common hereditary retinal degeneration, with a prevalence of 1 in 3,000 to 1 in 5,000 people.
- Harrison's Principles of Internal Medicine 22E, p. 275
- Kanski's Clinical Ophthalmology 10th Edition
Genetics and Inheritance
RP can occur sporadically or in several inheritance patterns:
| Pattern | Notes |
|---|---|
| Autosomal dominant (AD) | Best prognosis; most commonly due to mutations in the rhodopsin (RHO) gene |
| Autosomal recessive (AR) | Intermediate severity |
| X-linked | Least common but most severe; 90% of cases caused by mutations in the RPGR gene (encoding a protein in the connecting cilium of rods); often leads to visual acuity of 6/60 or less by the fifth decade |
| Sporadic | No affected family members identified; may be any of the above patterns with incomplete penetrance |
Mutations in more than 100 gene loci are known to cause non-syndromic RP, affecting:
- The phototransduction cascade (e.g., rhodopsin)
- The retinoid cycle
- Photoreceptor structure (e.g., peripherin, a glycoprotein in photoreceptor outer segments)
- Other biological functions of photoreceptors and RPE (e.g., RHO, USH2A, RPGR, EYS genes)
Despite these advances, about half of all affected individuals have no identified molecular genetic abnormality.
- Kanski's Clinical Ophthalmology 10th Edition
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Pathophysiology
Rods and cones die by apoptosis (cone death is partly mediated by RIP kinase activation). As rods are lost first:
- Early night blindness and constricted visual fields develop
- Loss of cones later impairs central visual acuity
The retinal atrophy is accompanied by:
- Constriction of retinal vessels
- Optic nerve head atrophy ("waxy pallor" of the disc)
- Accumulation of retinal pigment around blood vessels - this pigment accumulation gives the disease its name
Classic Triad of Fundus Findings
The hallmark diagnostic triad on fundoscopy:
- Bone-spicule pigmentation - irregular black deposits of clumped pigment in the peripheral retina resembling the spicules of cancellous bone
- Arteriolar attenuation - narrowing of retinal blood vessels
- "Waxy" disc pallor - pale optic disc
Here is what retinitis pigmentosa looks like on fundoscopy:
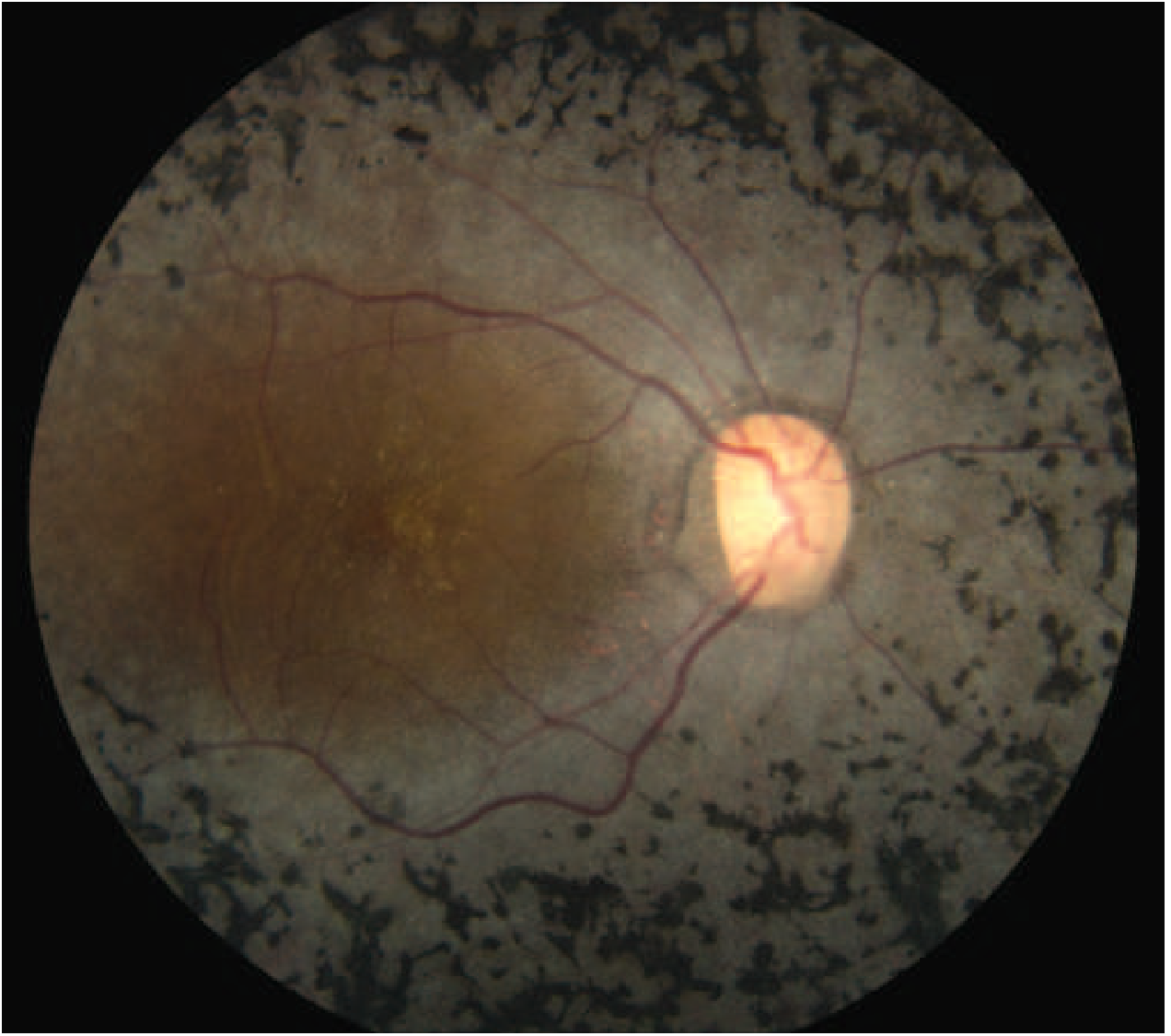
Retinitis pigmentosa showing the classic "bone spicule" black pigment deposits scattered throughout the peripheral retina, markedly attenuated arterioles, and a pale optic disc, with relative sparing of central (macular) vision.
Symptoms
| Symptom | Timing |
|---|---|
| Nyctalopia (night blindness) + dark adaptation difficulty | Usually the first symptom |
| Peripheral visual field loss (ring scotoma) | Early-to-mid disease |
| Photopsia (perceived flashes) | Not uncommon |
| Reduced central vision | Later feature (earlier if posterior subcapsular cataract develops) |
Signs
- Bilateral mid-peripheral intraretinal perivascular "bone-spicule" changes
- RPE atrophy
- Arteriolar narrowing
- Macular involvement: atrophy, epiretinal membrane (ERM), or cystoid macular oedema (CMO) in some patients
- Myopia is common
Complications: Posterior subcapsular cataract (common in all forms), open-angle glaucoma (~3%), posterior vitreous detachment, optic disc drusen.
Investigations
| Test | Finding |
|---|---|
| Full-field ERG | Most sensitive diagnostic test; reduced scotopic (rod) and combined responses early; eventually extinguished |
| Fundus autofluorescence (FAF) | Abnormal perimacular ring of hyper-autofluorescence; diffuse patchy hypo-autofluorescence mid-periphery; distinguishes RP from normal fundus in ~95% of cases |
| OCT | Detects macular oedema (~15% of patients) and macular atrophy (~40%); prognostic value |
| Visual field (perimetry) | Mid-peripheral scotomas that gradually coalesce; "tunnel vision" in advanced disease |
| Dark adaptation test | Prolonged; useful in equivocal early cases |
| Genetic analysis | Identifies the specific mutation; guides genetic counselling and trial eligibility |
Associated Syndromes ("Syndromic RP")
In 20-30% of cases, RP is part of a systemic syndrome (usually AR or mitochondrial inheritance):
- Usher syndrome - RP + sensorineural hearing loss
- Bardet-Biedl syndrome - RP + obesity, polydactyly, renal anomalies, intellectual disability
- Refsum disease - RP + peripheral neuropathy + ataxia (due to phytanic acid accumulation)
- Kearns-Sayre syndrome - RP + progressive external ophthalmoplegia + cardiac conduction defects (mitochondrial)
- Bassen-Kornzweig disease (abetalipoproteinemia)
- Olivopontocerebellar degeneration
- Leber congenital amaurosis - a severe early-onset form (~5% of RP cases)
Drugs That Mimic RP (Toxic Retinopathy)
- Chloroquine / Hydroxychloroquine - can cause a bull's-eye maculopathy resembling RP; patients on long-term hydroxychloroquine need regular OCT monitoring
- Phenothiazines (especially thioridazine)
Prognosis
- AD form: best prognosis
- AR form: intermediate severity
- X-linked form: most severe - central vision often 6/60 or worse by the fifth decade
Management
There is currently no curative treatment for most forms of RP, but several approaches exist:
- Gene therapy (Luxturna/voretigene neparvovec): FDA-approved for patients with biallelic RPE65 mutations; shown to improve visual function. A 2026 meta-analysis (PMID: 41773775) confirms efficacy and safety of gene therapy for RPGR gene-associated X-linked RP.
- Annual follow-up is recommended to detect treatable complications (cataract, CMO)
- CMO can be treated with oral carbonic anhydrase inhibitors
- Cataract surgery when visually significant
- Genetic counselling for family members
- Low vision aids and rehabilitation
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 34
- Kanski's Clinical Ophthalmology: A Systematic Approach, 10th Edition
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Recent evidence: gene therapy for RPGR-associated X-linked RP - Yang et al., 2026 (PMID: 41773775)
This is a shared conversation. Sign in to Orris to start your own chat.