Why in liver failure spironolactone is used and not other diuretics like thiazides and loop diuretics
spironolactone liver failure ascites mechanism why not loop diuretics thiazides
Why Spironolactone - Not Thiazides or Loop Diuretics - in Liver Failure
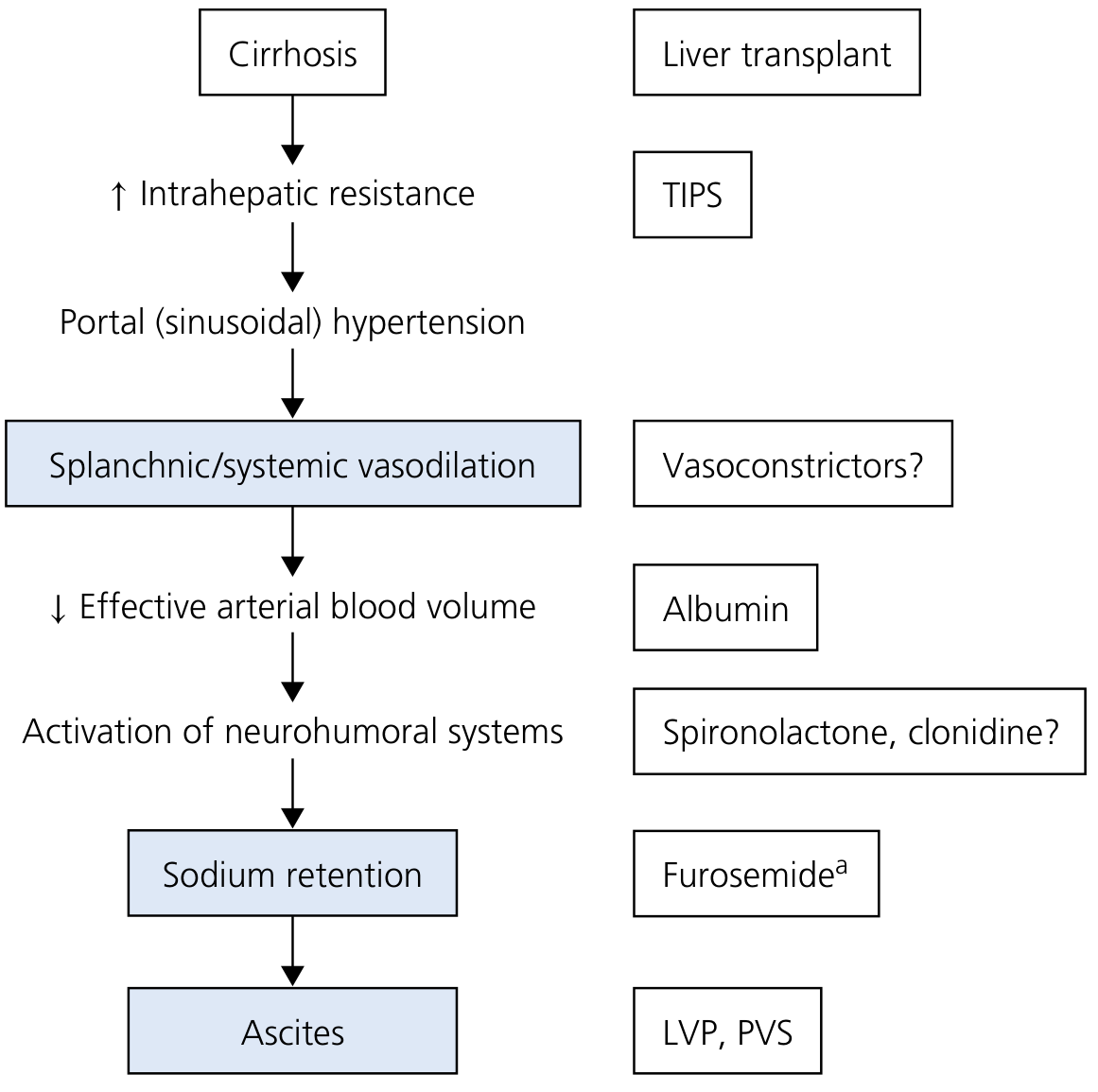
Step 1: Understand the Pathophysiology
- Increased production - activated RAAS drives the adrenal gland to make more aldosterone
- Decreased hepatic degradation - the failed liver cannot metabolize aldosterone at normal rates
Step 2: Why Spironolactone Works
"Because hyperaldosteronism plays a key role in renal sodium retention in cirrhosis, anti-mineralocorticoids (particularly spironolactone or eplerenone) represent the first line of treatment in the management of cirrhotic ascites."
Step 3: Why Loop Diuretics (e.g., Furosemide) Fail Alone
"When furosemide is used alone, sodium not reabsorbed in the loop of Henle is avidly absorbed in the distal and collecting tubules as a result of the hyperaldosteronism characteristic of most cirrhotic patients with ascites."
- Yamada's Textbook of Gastroenterology
- Hypokalemia - worsens hepatic encephalopathy (K+ depletion promotes ammonia production and renal ammonia generation)
- Metabolic alkalosis - further impairs brain function in encephalopathy
- Volume depletion - triggers hepatorenal syndrome (HRS), one of the most feared complications; only ~500 mL/day can safely be mobilized from ascites without causing circulatory dysfunction
Step 4: Why Thiazides Are Not Used
- Thiazides carry a high risk of hyponatremia - already a major problem in advanced cirrhosis (dilutional hyponatremia from ADH activation)
- Thiazides also cause hypokalemia, again risking encephalopathy
- There is no evidence they offer any advantage over the spironolactone/furosemide combination
- As noted by Yamada's Gastroenterology: "There is no evidence that other diuretics, such as metolazone, thiazides, or other loop diuretics such as bumetanide or torsemide, offer an advantage over spironolactone and furosemide."
Practical Clinical Protocol
| Situation | First-line |
|---|---|
| Mild-moderate ascites | Spironolactone 100 mg/day alone |
| Inadequate response | Add furosemide 40 mg/day (ratio kept at 100:40 to maintain normokalemia) |
| Titrate up to maximum | Spironolactone 400 mg + Furosemide 160 mg |
| Painful gynecomastia | Switch to amiloride (another K+-sparing diuretic, though less effective) |
- Renal impairment from intravascular depletion (~25% of patients)
- Hyponatremia (~28%)
- Hepatic encephalopathy (~26%)
- Hyperkalemia (especially with spironolactone in renal impairment)
Summary Table
| Diuretic | Site of action | Problem in liver failure |
|---|---|---|
| Spironolactone | Collecting duct (blocks aldosterone) | Preferred - targets the root cause |
| Furosemide | Loop of Henle | Sodium reabsorbed downstream by aldosterone; causes hypokalemia/HRS |
| Thiazides | Distal convoluted tubule | Same bypass problem; causes hyponatremia/hypokalemia |
| Amiloride | Collecting duct (blocks ENaC directly) | Alternative if gynecomastia; less natriuretic effect |
Action of loop diuretics in the blood electrolytes levels
loop diuretics electrolyte effects blood Na K Mg Ca mechanism
Effects of Loop Diuretics on Blood Electrolytes
The Root Mechanism
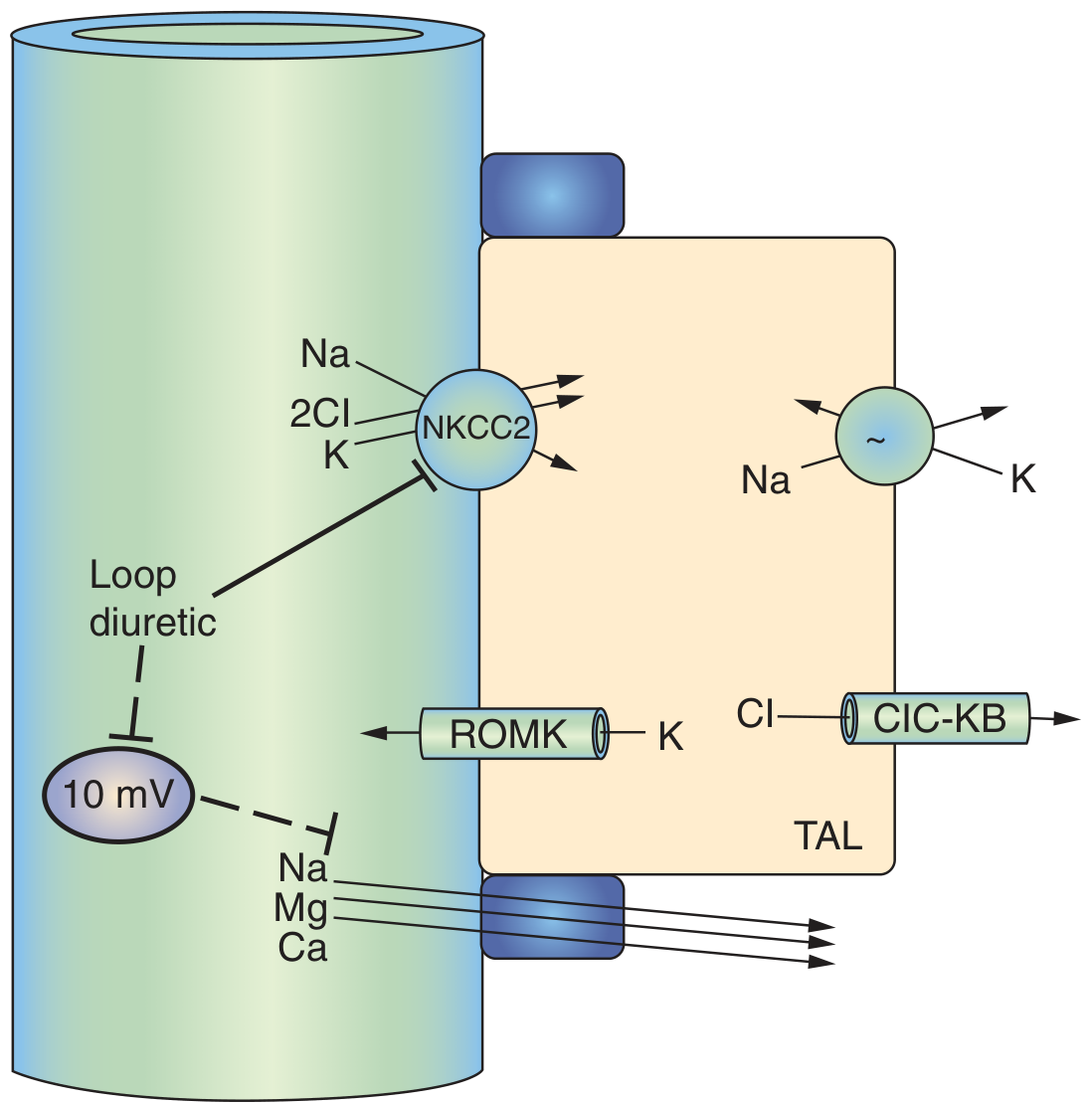
1. Hyponatremia (↓ Na⁺)
- NKCC2 blockade directly prevents Na⁺ reabsorption in the TAL, which accounts for ~25% of all filtered sodium
- The increased distal sodium delivery overwhelms downstream reabsorption capacity
- Volume depletion from Na⁺ loss stimulates ADH release (non-osmotic), causing water retention - this dilutes serum Na⁺ further
- Secondary RAAS activation causes more renin/aldosterone release, driving more Na⁺ retention (partially compensatory), but at the expense of K⁺
2. Hypokalemia (↓ K⁺) - the most clinically important electrolyte effect
- NKCC2 blockade means more Na⁺ is delivered to the principal cells of the collecting duct
- The Na⁺/K⁺-ATPase in principal cells reabsorbs this extra Na⁺ by exchanging it for K⁺ secretion into the lumen - K⁺ is lost in urine
- Loop diuretics stimulate renin secretion directly via the macula densa (by blocking NaCl entry into macula densa cells, reducing TGF-mediated feedback)
- As Brenner & Rector states: "Loop diuretics also stimulate renin secretion, both in the short term and long term... a major component is from direct effects on the macula densa"
- More renin → more angiotensin II → more aldosterone → aldosterone further amplifies K⁺ secretion at the collecting duct
3. Hypochloremia (↓ Cl⁻)
- NKCC2 directly transports 2 Cl⁻ per cycle - blocking it massively increases urinary Cl⁻ loss
- Cl⁻ is lost in urine alongside Na⁺ and K⁺
4. Metabolic Alkalosis (↑ HCO₃⁻, ↑ pH)
- Volume contraction alkalosis - loss of Cl⁻-rich fluid contracts the ECF volume; a fixed amount of HCO₃⁻ is now dissolved in a smaller volume → "contraction alkalosis"
- Hypokalemia drives alkalosis - K⁺ depletion causes H⁺ to shift into cells in exchange for K⁺, lowering extracellular [H⁺] → alkalosis
- Aldosterone effect - secondary hyperaldosteronism drives H⁺ secretion by intercalated cells of the collecting duct, generating more HCO₃⁻
5. Hypomagnesemia (↓ Mg²⁺)
"Loop diuretics eliminate this potential difference and can therefore increase calcium and magnesium excretion."
6. Hypocalcemia (↓ Ca²⁺) - key distinction from thiazides
- Ca²⁺ (~20% of filtered load) is reabsorbed passively via paracellular route in the TAL, driven by the +10 mV transepithelial voltage
- Loop diuretics abolish this voltage → Ca²⁺ lost in urine
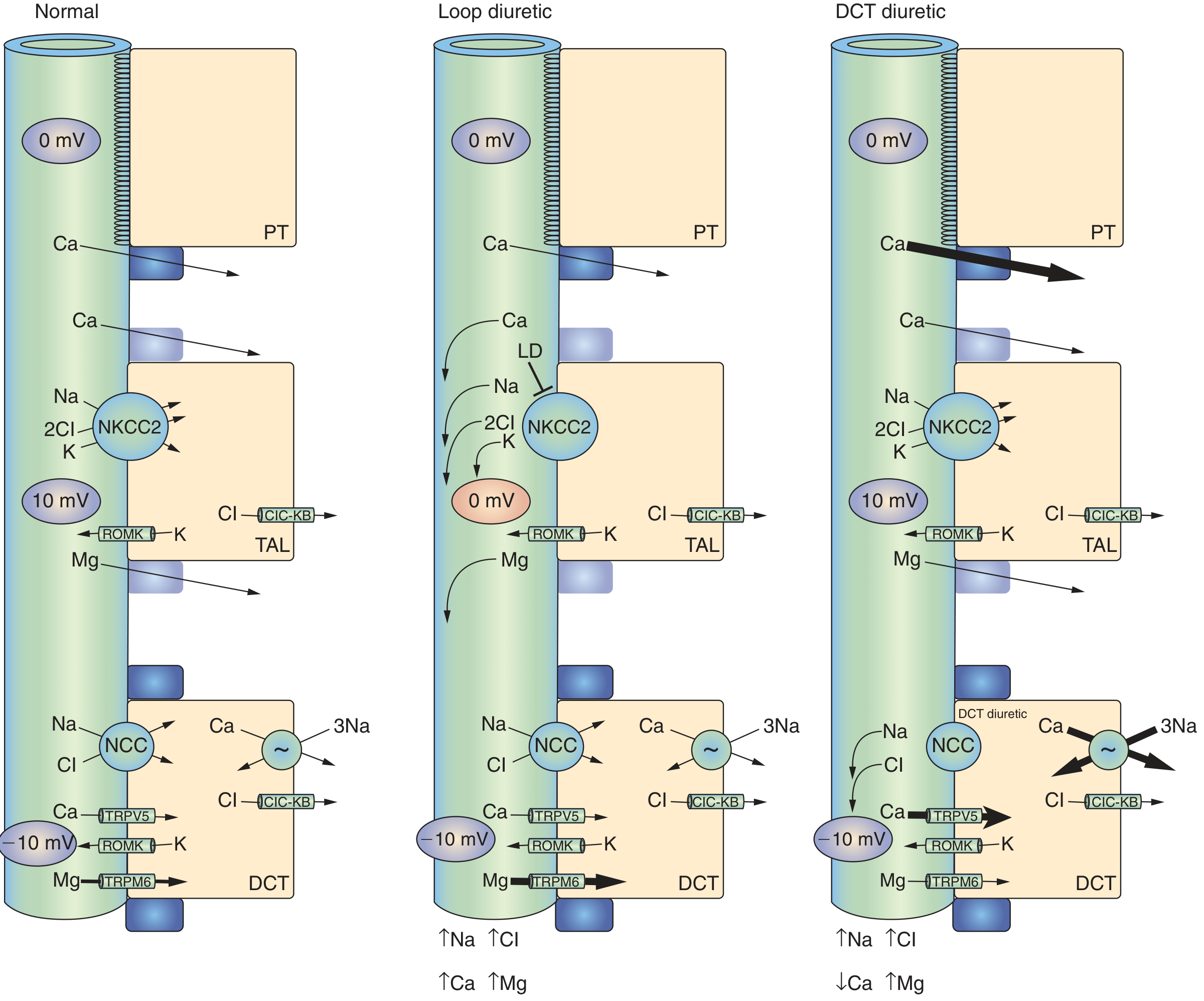
- Loop diuretics → hypocalcemia → used clinically to treat hypercalcemia
- Thiazides → hypercalcemia → preferred in patients with osteoporosis or calcium stones
7. Hyperuricemia (↑ Uric acid) - bonus effect
- Acutely, loop diuretics increase uric acid excretion
- Chronically, volume depletion enhances proximal tubular uric acid reabsorption (via OAT transporters), raising serum uric acid
- Can precipitate gout in susceptible patients
Summary Table
| Electrolyte | Blood Level | Key Mechanism |
|---|---|---|
| Na⁺ | ↓ (hyponatremia) | Direct TAL blockade + ADH-driven water retention |
| K⁺ | ↓ (hypokalemia) | ↑ distal Na⁺ delivery + secondary hyperaldosteronism |
| Cl⁻ | ↓ (hypochloremia) | NKCC2 directly transports 2Cl⁻ |
| HCO₃⁻/pH | ↑ (metabolic alkalosis) | Volume contraction + hypokalemia + aldosterone |
| Mg²⁺ | ↓ (hypomagnesemia) | Loss of +10mV transepithelial voltage in TAL |
| Ca²⁺ | ↓ (hypocalcemia) | Loss of +10mV transepithelial voltage in TAL |
| Uric acid | ↑ (hyperuricemia) | Chronic proximal tubular reabsorption via volume contraction |
- Hypokalemia and hypomagnesemia are interlinked - you often cannot correct K⁺ without first repleting Mg²⁺, because low Mg²⁺ impairs renal K⁺ retention
- The metabolic alkalosis from loop diuretics is Cl⁻-responsive (corrects with volume and KCl repletion)
- Furosemide's calciuric effect is exploited therapeutically in hypercalcemic crisis (IV furosemide + IV saline drives urinary Ca²⁺ loss)
How does metformin causes B12 deficiency
metformin vitamin B12 deficiency mechanism calcium intrinsic factor ileum
metformin vitamin B12 deficiency mechanism
How Metformin Causes Vitamin B12 Deficiency
Normal B12 Absorption (Brief Review)
- Stomach - Dietary B12 (cobalamin) is released from food proteins by pepsin and gastric acid, then binds to a salivary protein called haptocorrin (R-protein)
- Duodenum - Pancreatic proteases release B12 from haptocorrin; free B12 then binds intrinsic factor (IF), secreted by gastric parietal cells
- Terminal ileum - The B12-IF complex binds to a specific receptor called cubilin on the surface of ileal epithelial cells. This step is calcium-dependent - Ca²⁺ is required for the complex to dock onto the receptor
- Endocytosis - The B12-IF-cubilin complex is internalized; within the cell, B12 binds transcobalamin II and enters the portal circulation
- Liver - B12 is stored in large amounts (enough for 3-5 years), and undergoes enterohepatic recirculation via bile
How Metformin Disrupts This
Mechanism 1: Calcium-Dependent Ileal Absorption Blockade (Primary Mechanism)
- Metformin is a biguanide with a hydrophobic tail that inserts into the hydrocarbon core of intestinal cell membranes
- This insertion imparts a net positive charge to the cell membrane surface
- The positively charged membrane repels Ca²⁺ ions at the ileal lumen-cell interface
- Without adequate Ca²⁺, the B12-IF complex cannot bind to the cubilin receptor on ileal epithelial cells
- The complex passes through the gut unabsorbed and is lost in stool
"Metformin → Vitamin B₁₂ → Impairs absorption" (drug-nutrient interaction table)
Mechanism 2: Gut Microbiome Alteration and Bacterial Overgrowth
- Metformin is known to significantly alter the gut microbiome - it promotes growth of certain bacteria (notably Akkermansia muciniphila and Bifidobacterium) and suppresses others
- Some of the altered bacterial species consume vitamin B12 for their own metabolic needs, reducing the amount available for absorption
- Additionally, bacterial overgrowth in the small intestine can compete for the B12-IF complex before it reaches the terminal ileum
- Metformin also slows intestinal transit (contributes to its GI side effects), which may prolong bacterial exposure to the B12-IF complex
Mechanism 3: Direct Structural Interference with the B12-IF Complex
- A third proposed mechanism suggests metformin may directly bind to the B12-IF complex, altering its structure
- A structurally modified complex would have reduced affinity for the cubilin receptor, even if calcium is available
- This mechanism is less well characterized than Mechanism 1
Why This Matters Clinically
- Dose-dependent - higher daily doses = greater risk
- Duration-dependent - longer treatment = greater cumulative depletion of hepatic stores
- Seen in up to 10-30% of long-term users in various studies
- Clinically silent early - hepatic B12 stores last 3-5 years, so deficiency can develop insidiously over years of treatment
Risk Factors for Greater Depletion
| Risk Factor | Why It Worsens B12 Depletion |
|---|---|
| High metformin dose (>1.5 g/day) | More membrane charge displacement |
| Duration >5 years | Hepatic stores eventually depleted |
| Vegetarian/vegan diet | Low dietary B12 intake to begin with |
| Concomitant PPI or H2 blocker use | PPIs reduce gastric acid → impair B12 release from food; together with metformin, up to 65% reduction in B12 absorption |
| Elderly patients | More atrophic gastritis, reduced IF secretion, reduced gastric acid |
| Pre-existing low B12 stores | Less buffer before clinical deficiency |
Summary of the Mechanism
Metformin (hydrophobic tail inserts into ileal cell membrane)
↓
Net positive charge on membrane surface
↓
Ca²⁺ ions repelled from ileal lumen-cell interface
↓
B12-Intrinsic Factor complex cannot bind cubilin receptor
↓
B12-IF complex not endocytosed → lost in stool
↓
Reduced circulating B12 → hepatic stores slowly depleted
↓
Megaloblastic changes + Peripheral/autonomic neuropathy
Management
- Routine monitoring: Check serum B12 every 1-2 years in long-term metformin users; use MMA levels if borderline
- Calcium supplementation: 1200 mg/day calcium carbonate can partially restore absorption
- B12 supplementation: Oral B12 1000 µg/day (works via passive diffusion, even without IF) or IM cyanocobalamin if severe deficiency
- Do not stop metformin for this reason alone - the cardiovascular and glycemic benefits far outweigh the risk when monitored and supplemented appropriately
List me the various names effects in medicine ex Macklin effect
famous named effects phenomena in medicine eponymous list clinical
named effects in medicine "Bohr effect" "Haldane effect" "Cushing reflex" "Starling law" "Bainbridge reflex" physiology eponymous effects list
Named Effects in Medicine - A Comprehensive List
RESPIRATORY
CARDIOVASCULAR
RENAL / FLUID
BLOOD / HEMATOLOGY
PHARMACOLOGY / TOXICOLOGY
NEUROLOGICAL / BRAIN
ENDOCRINE / METABOLIC
PULMONARY / CRITICAL CARE
GASTROENTEROLOGY / SURGERY
IMMUNOLOGY / ONCOLOGY
QUICK-REFERENCE SUMMARY TABLE
| Effect | System | One-Line Summary |
|---|---|---|
| Macklin | Respiratory | Alveolar rupture → pneumomediastinum via bronchovascular sheath |
| Bohr | Respiratory | ↑CO₂/↑H⁺ → right shift of ODC → O₂ unloading |
| Haldane | Respiratory | Deoxy-Hb carries more CO₂ than oxy-Hb |
| Euler-Liljestrand | Respiratory | Hypoxia → local pulmonary vasoconstriction → V/Q matching |
| Frank-Starling | Cardiac | ↑Preload → ↑Stroke volume |
| Bowditch/Treppe | Cardiac | ↑Heart rate → ↑Contractility |
| Anrep | Cardiac | ↑Afterload → slow recovery of contractility |
| Cushing | Neuro/Cardiac | ↑ICP → hypertension + bradycardia + irregular breathing |
| Bainbridge | Cardiac | ↑Atrial pressure → tachycardia |
| Bezold-Jarisch | Cardiac | Cardiac C-fiber stimulation → bradycardia + hypotension |
| Windkessel | Vascular | Aortic elastic recoil maintains diastolic pressure |
| Doppler | Vascular | Wave frequency shift used in blood flow measurement |
| Starling Forces | Renal | Four forces governing capillary fluid exchange |
| Warburg | Oncology | Cancer cells use aerobic glycolysis (↑PET uptake) |
| Abscopal | Oncology | Local RT causes systemic tumor regression via immunity |
| Prozone | Immunology | Antigen excess → false negative serology |
| First-Pass | Pharmacology | Hepatic pre-systemic drug metabolism |
| Tachyphylaxis | Pharmacology | Rapid drug tolerance with repeated doses |
| Somogyi | Endocrine | Nocturnal hypoglycemia → morning hyperglycemia rebound |
| Dawn Phenomenon | Endocrine | GH/cortisol surge → morning hyperglycemia |
| Spreading Depression | Neuro | Wave of depolarization across cortex → migraine aura |
| Kindling | Neuro | Repeated sub-threshold stimulation → seizures |
| 2,3-BPG Effect | Hematology | ↑2,3-BPG → right shift ODC → better O₂ delivery in hypoxia |
| Courvoisier | GI | Palpable painless gallbladder = malignant obstruction |
Create a study guide PDF of named effects organized by system
Named Effects in Medicine
PDF Document · PDF
- Cover page - title, system overview, total count (38 effects across 8 systems)
- Table of contents - all effects listed by system with sub-entries
- 8 colour-coded system sections, each with a distinct banner colour:
- Respiratory (7 effects) - Macklin, Bohr, Haldane, Euler-Liljestrand, Pendelluft, Paul Bert, Bernoulli/Venturi
- Cardiovascular (8 effects) - Frank-Starling, Bowditch, Anrep, Cushing, Bainbridge, Bezold-Jarisch, Windkessel, Fick
- Renal & Fluid (4 effects) - Starling Forces, Tubuloglomerular Feedback, Gibbs-Donnan, Countercurrent Multiplication
- Haematology (3 effects) - 2,3-BPG, Prozone, Rouleaux
- Neurology (4 effects) - Leão spreading depression, Kindling, Mass effect, Doppler
- Pharmacology & Toxicology (4 effects) - First-pass, Tachyphylaxis, Warburg, Crabtree
- Endocrine & Metabolic (3 effects) - Somogyi, Dawn phenomenon, Staub-Traugott
- Gastroenterology & Surgery, Immunology & Oncology
- Each effect includes: name, eponymous credit, mechanism description, and a clinical pearl in orange
- Final quick-reference table - all 38 effects in a single scannable table with one-line summaries
What is brisk diuretics
brisk diuresis medical term meaning clinical use
"brisk diuresis" clinical meaning urine output rate mL/kg/hr
Toxaemia meaning
toxaemia meaning definition medicine pregnancy preeclampsia sepsis
Toxaemia - Meaning
1. General / Older Meaning - Blood Poisoning
- Septicaemia - bacteria themselves in the blood
- Sepsis - the body's dysregulated response to infection with systemic organ effects
- Endotoxaemia - specifically gram-negative bacterial lipopolysaccharide (LPS/endotoxin) in the blood
2. Obstetric Meaning - Toxaemia of Pregnancy (Pre-eclampsia)
What is Pre-eclampsia?
- New-onset hypertension (BP ≥ 140/90 mmHg) after 20 weeks' gestation
- Proteinuria (≥ 300 mg/24 hours, or protein:creatinine ratio ≥ 30 mg/mmol)
- And/or signs of end-organ damage (renal, hepatic, neurological, haematological, or uteroplacental)
The Toxaemia → Pre-eclampsia → Eclampsia Spectrum
| Stage | Defining Features |
|---|---|
| Gestational hypertension | High BP after 20 weeks, no proteinuria, no organ damage |
| Pre-eclampsia (Toxaemia) | High BP + proteinuria and/or organ damage after 20 weeks |
| Severe pre-eclampsia | BP ≥ 160/110, severe headache, visual disturbance, pulmonary oedema, oliguria, epigastric pain |
| Eclampsia | Pre-eclampsia + generalised tonic-clonic seizures |
| HELLP syndrome | Haemolysis + Elevated Liver enzymes + Low Platelets - a severe variant |
Why Does Pre-eclampsia Happen? (Pathophysiology in Brief)
- Shallow trophoblast invasion of spiral arteries in early pregnancy - the arteries fail to remodel into wide, low-resistance vessels
- Placental ischaemia results, causing the placenta to release anti-angiogenic factors into maternal circulation, especially:
- sFlt-1 (soluble FMS-like tyrosine kinase-1) - traps VEGF and PlGF, reducing their vasodilatory effects
- sEng (soluble endoglin) - blocks TGF-β signalling
- These factors cause widespread maternal endothelial dysfunction → vasoconstriction → hypertension, proteinuria (glomerular endotheliosis), oedema, and multi-organ damage
Risk Factors for Pre-eclampsia (Toxaemia)
| Risk Factor | Relative Risk |
|---|---|
| Prior pre-eclampsia | ~8× |
| Chronic hypertension | ~5× |
| Antiphospholipid syndrome | ~3× |
| Nulliparity (first pregnancy) | ~2× |
| Multiple gestation (twins/triplets) | ↑ |
| Diabetes, obesity, renal disease | ↑ |
Bottom Line
"Toxaemia" = an old term with two uses:
- Broadly: toxins/bacteria in the blood (now called sepsis/septicaemia)
- In obstetrics: the old name for pre-eclampsia - hypertension + proteinuria after 20 weeks of pregnancy, caused by placental dysfunction and maternal endothelial injury