Pathogenesis of septic shock
Reading File
Finding Sources
Finding Sources
Searching PubMed
"septic shock"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have everything needed for a comprehensive, well-illustrated answer. Here is the full response:
Pathogenesis of Septic Shock
Septic shock is defined as sepsis-induced circulatory failure causing persistent hypotension despite adequate fluid resuscitation, requiring vasopressors, and associated with a serum lactate >2 mmol/L. Mortality remains 20-40% despite modern critical care.
Triggering Organisms
Septic shock is most frequently triggered by gram-positive bacteria, followed by gram-negative bacteria and fungi. Emerging viral pathogens (e.g., SARS-CoV-2) can also cause sepsis. The ability of diverse organisms to produce the same syndrome reflects the fact that many microbial constituents - endotoxin (LPS), staphylococcal enterotoxins, streptococcal M protein - all converge on the same host pathways.
The Central Concept
Sepsis represents a failure of normal containment mechanisms that have evolved to limit infection to its site of origin. When the local inflammatory response becomes systemic and dysregulated, it injures normal tissues remote from the infection site. The end result is a complex interplay of four major pathophysiological arms:
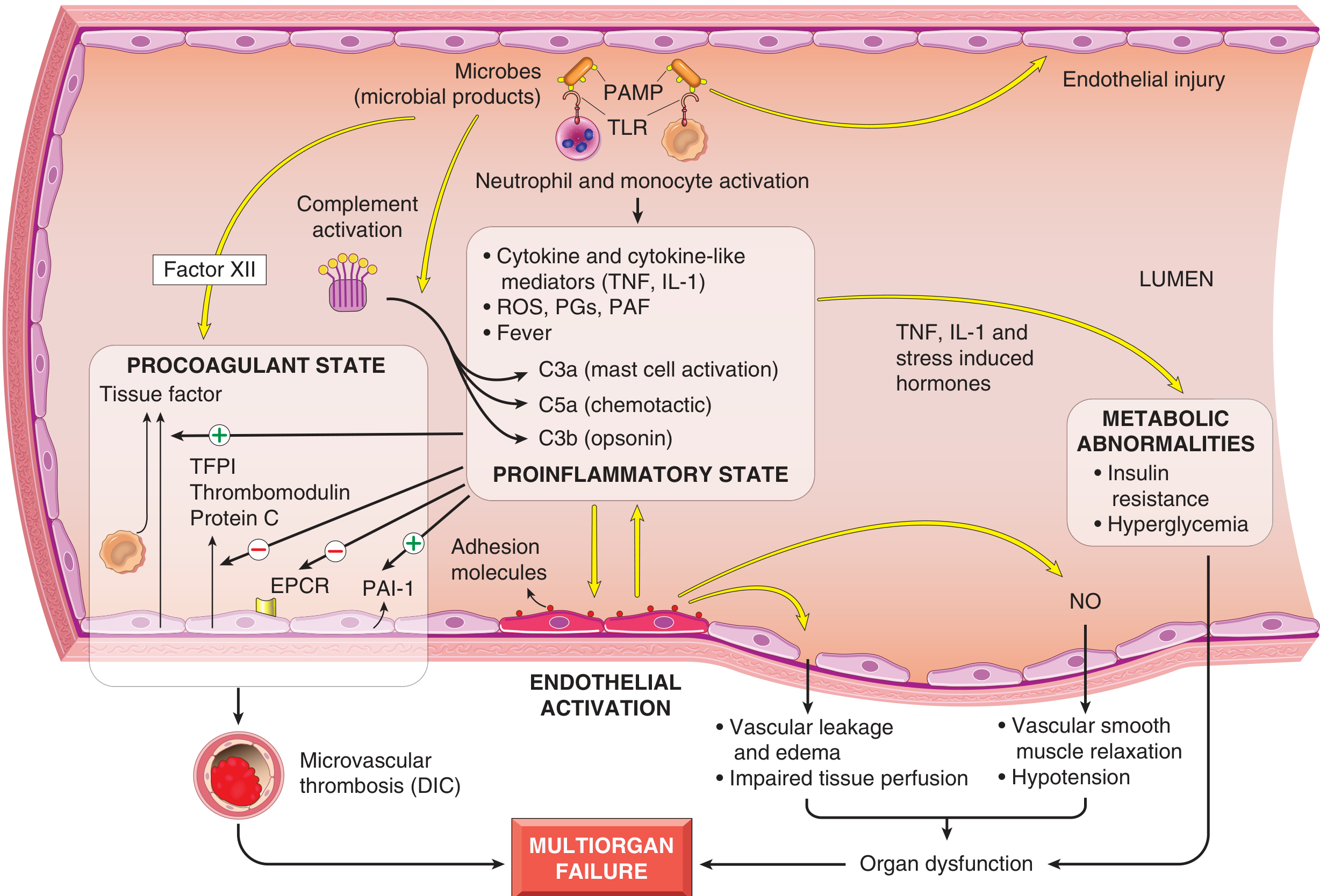
Fig. 3.19 - Major pathogenic pathways in septic shock. Microbial products activate endothelial cells and innate immune elements, initiating cascades leading to a procoagulant, proinflammatory state and multiorgan failure.
1. Inflammatory and Counter-Inflammatory Responses
Pattern Recognition and Initial Activation
Microbial cell wall constituents (PAMPs - pathogen-associated molecular patterns) engage three main classes of receptors on macrophages, neutrophils, dendritic cells, and endothelial cells:
- Toll-like receptors (TLRs) - recognize PAMPs and DAMPs (damage-associated molecular patterns from injured host cells), signaling primarily via NF-kB nuclear translocation
- G-protein-coupled receptors - detect bacterial peptides (e.g., fMLP)
- C-type lectin receptors (Dectins) - detect fungal cell wall components
Cytokine Storm
NF-kB activation drives massive upregulation of:
- TNF and IL-1 - the primary proximal cytokines of sepsis; amplify the entire downstream cascade
- IL-12, IL-18, IFN-γ - drive Th1 polarization and macrophage activation
- HMGB1 (High-Mobility Group Box 1 protein) - a late mediator that sustains inflammation
- Acute-phase reactants - CRP and procalcitonin rise as markers of the response
Reactive oxygen species (ROS), prostaglandins, and platelet-activating factor (PAF) are also elaborated.
Complement Activation
The complement cascade is activated both directly by microbial components and indirectly through plasmin, generating:
- C3a, C5a - anaphylatoxins causing mast cell degranulation and further cytokine release
- C5a - potent chemotactic fragment recruiting neutrophils
- C3b - opsonin that enhances phagocytosis
Counter-Inflammatory (Immunosuppressive) Phase
The hyperinflammatory state paradoxically triggers counterregulatory mechanisms:
- Shift from pro-inflammatory Th1 to anti-inflammatory Th2 cytokines
- Production of soluble TNF receptor, IL-1 receptor antagonist, and IL-10
- Lymphocyte apoptosis (especially CD4+ T cells and B cells)
- Immunosuppressive effects of apoptotic cells on surviving innate immune cells
As a result, septic patients oscillate between hyperinflammatory and immunosuppressed states during their clinical course. The intensity of each phase depends on host genetics, comorbidities, and pathogen virulence/burden. This biphasic pattern explains why anti-TNF strategies have failed clinically - blocking inflammation during the immunosuppressive phase worsens outcomes.
2. Endothelial Activation and Injury
Endothelial injury is a central event linking the inflammatory cascade to the clinical manifestations of shock:
- Vascular leakage: Cytokines (particularly TNF and IL-1) loosen endothelial tight junctions, causing protein-rich edema fluid to accumulate throughout the body - impairing both nutrient delivery and waste removal
- Tissue edema is paradoxically worsened by IV fluid resuscitation attempts
- Nitric oxide (NO) overproduction: Inflammatory cytokines upregulate inducible NOS (iNOS) in endothelial and smooth muscle cells, producing massive NO release → vascular smooth muscle relaxation → systemic vasodilation → refractory hypotension
- Adhesion molecule upregulation: ICAM-1, E-selectin, etc. promote neutrophil margination and tissue infiltration
- Microvascular dysfunction: Heterogeneity of capillary flow, loss of autoregulation - a mismatch between oxygen delivery and tissue oxygen demand even when global cardiac output appears preserved
3. Induction of a Procoagulant State (DIC)
Sepsis creates a profound coagulopathy affecting up to 50% of septic patients, sufficient to cause disseminated intravascular coagulation (DIC):
| Factor | Direction in Sepsis | Mechanism |
|---|---|---|
| Tissue factor (TF) on monocytes/endothelium | ↑ Increased | Driven by TNF, IL-1 |
| Thrombomodulin | ↓ Decreased | Endothelial downregulation |
| TFPI (Tissue Factor Pathway Inhibitor) | ↓ Decreased | Endothelial downregulation |
| Protein C / EPCR | ↓ Decreased | Reduced endothelial expression |
| PAI-1 (Plasminogen Activator Inhibitor-1) | ↑ Increased | Suppresses fibrinolysis |
Additional pro-coagulant mechanisms:
- Factor XII (Hageman factor) is directly activated by microbial components (intrinsic pathway)
- Neutrophil extracellular traps (NETs) stimulate both intrinsic and extrinsic coagulation pathways
- Vascular stasis from tissue edema diminishes washout of activated clotting factors
The combined effect: systemic thrombin activation and widespread fibrin-rich microthrombi in small vessels, further compromising tissue perfusion. In full-blown DIC, consumption of clotting factors and platelets leads to the paradox of simultaneous thrombosis and hemorrhage.
4. Metabolic Abnormalities
Sepsis induces a characteristic metabolic derangement driven by TNF, IL-1, and stress-induced hormones (glucagon, growth hormone, glucocorticoids, catecholamines):
- Insulin resistance - proinflammatory cytokines suppress insulin release and impair GLUT-4 surface expression in liver and peripheral tissues
- Hyperglycemia - from gluconeogenesis plus insulin resistance; further suppresses neutrophil bactericidal function and upregulates endothelial adhesion molecules (vicious cycle)
- Hypertriglyceridemia and lactic acidosis - from increased lactate production
- Adrenal insufficiency - initial glucocorticoid surge may be followed by functional adrenal insufficiency, either from suppression of adrenal synthetic capacity or frank adrenal necrosis from DIC (Waterhouse-Friderichsen syndrome)
- Mitochondrial dysfunction - oxidative stress impairs oxidative phosphorylation, causing cytopathic hypoxia (cells cannot use oxygen even when it is delivered)
5. Multiorgan Dysfunction and Failure
All four arms converge to produce multiorgan failure (MOF):
Hypotension + Tissue edema + Microvascular thrombosis
↓
Reduced O₂ / nutrient delivery + Mitochondrial dysfunction
↓
Cellular hypoxia → Organ dysfunction
- Kidneys - septic AKI (occurs in up to 2/3 of septic patients); oliguria
- Lungs - ARDS from increased vascular permeability and endothelial injury
- Heart - myocardial depression from cytokines and ischemia; reduced contractility
- Liver - impaired hepatic perfusion and metabolic failure
Superantigens: A Special Case
Bacterial superantigens (e.g., staphylococcal toxic shock toxin-1, TSST-1) cause a clinically similar syndrome via a distinct mechanism: they are polyclonal T-cell activators that bypass normal antigen presentation, directly crosslinking MHC class II and TCR V-beta regions. Up to 20% of all T cells are simultaneously activated, releasing massive cytokine quantities - producing the toxic shock syndrome with diffuse rash, vasodilation, hypotension, and potential death.
Stages of Shock (Common Final Pathway)
| Stage | Features |
|---|---|
| 1. Non-progressive (compensated) | Baroreceptors, catecholamines, ADH, RAAS activated; tachycardia, peripheral vasoconstriction, renal fluid conservation; vital organ perfusion maintained |
| 2. Progressive | Widespread tissue hypoxia; anaerobic metabolism; metabolic acidosis; myocardial depression; worsening circulatory failure |
| 3. Irreversible | Cellular injury so severe that survival is impossible even if hemodynamics are restored; multiorgan failure and death |
Note: In septic shock, the early phase may show warm, flushed skin (from peripheral vasodilation via NO), unlike cardiogenic/hypovolemic shock which presents with cold, clammy, pale skin.
Why Targeted Therapies Have Failed
The complexity of sepsis pathogenesis explains a critical clinical lesson: single-mediator antagonism (anti-TNF, anti-IL-1, activated protein C, etc.) has repeatedly failed in clinical trials and even caused harm in some cases. This is because:
- Multiple redundant pathways exist
- Blocking inflammation during the immunosuppressive phase worsens outcomes
- Individual patients may be at very different points in the pro- vs. anti-inflammatory oscillation
Current standard of care remains source control (antibiotics targeting the underlying infection), IV fluids, vasopressors (norepinephrine first-line), and supplemental oxygen.
Sources:
- Robbins & Kumar Basic Pathology, pp. 97-98 (Fig. 3.19 - Pathogenesis of Septic Shock)
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 134-136 (Fig. 4.20)
This is a shared conversation. Sign in to Orris to start your own chat.