Congenital hyperbilirubinemia causes and explaination and syndromes
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Searching Images
bilirubin metabolism pathway conjugation jaundice diagram
I now have comprehensive information from multiple authoritative textbooks. Let me compile the complete answer.
Congenital Hyperbilirubinemia: Causes, Mechanisms & Syndromes
Congenital (hereditary) hyperbilirubinemia arises from genetic defects in bilirubin uptake, conjugation, or excretion. They are classified based on whether the excess bilirubin is unconjugated (indirect) or conjugated (direct).
Bilirubin Metabolism — Brief Overview
Bilirubin is produced from heme degradation → transported to the liver bound to albumin → taken up by hepatocytes via OATP1B1/OATP1B3 → conjugated with glucuronic acid by UGT1A1 → excreted into bile via MRP2 (canalicular transporter).
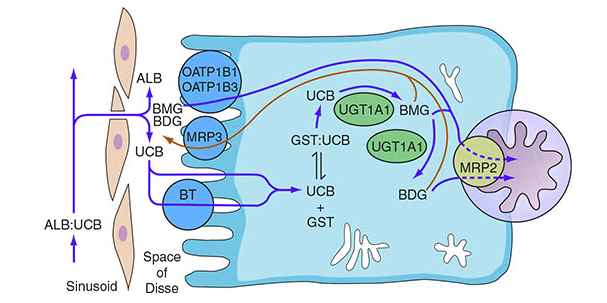
Bilirubin transport from sinusoid through hepatocyte to bile canaliculus — Harrison's Principles of Internal Medicine
Classification
| Type | Defect | Examples |
|---|---|---|
| Unconjugated | Conjugation failure (↓UGT1A1) | Gilbert, Crigler-Najjar I & II |
| Conjugated | Excretion/transport failure | Dubin-Johnson, Rotor |
| Cholestatic | Bile formation/export | PFIC types 1–3 |
UNCONJUGATED HYPERBILIRUBINEMIA SYNDROMES
1. Gilbert Syndrome
- Most common inherited cause; affects 3–12% of the population (male predominance 1.5–7:1)
- Gene: Promoter region polymorphism in UGT1A1 (TA repeat insertion: UGT1A1*28) → reduced transcription factor binding → UGT1A1 activity reduced to 10–33% of normal
- Inheritance: Autosomal recessive (some classify as complex haplotype trait)
- Bilirubin: Mild unconjugated hyperbilirubinemia, usually < 4–6 mg/dL, fluctuates
- Triggers: Fasting, stress, alcohol, concurrent illness, exercise
- Clinical: Essentially harmless — no liver damage, no kernicterus risk; may actually be cardioprotective and anti-inflammatory due to bilirubin's antioxidant properties
- Labs: All liver function tests otherwise normal
- Treatment: None required
2. Crigler-Najjar Syndrome Type I
- Gene: Complete loss-of-function mutations (nonsense, frameshift) in UGT1A1 → total absence of bilirubin UDP-glucuronosyltransferase activity
- Inheritance: Autosomal recessive
- Bilirubin: Severely elevated unconjugated bilirubin > 20 mg/dL (often 18–45 mg/dL)
- Clinical:
- Severe neonatal jaundice
- Kernicterus (bilirubin encephalopathy) — bilirubin crosses the blood-brain barrier (only unconjugated can) → brain damage, mental retardation
- Frequently fatal within the first 15 months of life without treatment
- Treatment:
- Phototherapy 10–12 hours/day (blue light converts bilirubin to a water-soluble isomer excreted in urine)
- Phenobarbital has NO effect (no UGT1A1 activity to induce)
- Liver transplantation is the definitive cure
3. Crigler-Najjar Syndrome Type II (Arias Syndrome)
- Gene: Nonsynonymous point mutations in UGT1A1 → residual activity ≤ 10% of normal (not total absence)
- Inheritance: Autosomal recessive
- Bilirubin: Elevated but usually < 20 mg/dL (range 6–25 mg/dL)
- Clinical:
- More benign than Type I; patients survive into adulthood
- Kernicterus is rare but can occur under metabolic stress (illness, surgery)
- Mild or no neurological impairment under normal conditions
- Treatment:
- Phenobarbital induces residual UGT1A1, reducing bilirubin by up to 75%
- Phototherapy if needed
Spectrum Summary: UGT1A1 Disorders
| Syndrome | UGT1A1 Activity | Bilirubin Level | Prognosis |
|---|---|---|---|
| Crigler-Najjar I | ~0% (absent) | 18–45 mg/dL | Fatal without Tx |
| Crigler-Najjar II | ≤10% | 6–25 mg/dL | Survives to adulthood |
| Gilbert Syndrome | 10–33% | Usually < 4 mg/dL | Benign |
CONJUGATED HYPERBILIRUBINEMIA SYNDROMES
4. Dubin-Johnson Syndrome
- Gene: Mutations in ABCC2 (encoding MRP2, multidrug resistance-associated protein 2) → impaired canalicular secretion of conjugated bilirubin into bile
- Inheritance: Autosomal recessive
- Bilirubin: Conjugated (direct) hyperbilirubinemia, typically 2–5 mg/dL (~60% direct-reacting); can worsen with estrogens (oral contraceptives), pregnancy, or infections
- Clinical:
- Asymptomatic jaundice, often intermittent
- Occasional hepatosplenomegaly
- Hallmark: Liver appears grossly black due to deposition of dark melanin-like pigment in hepatocytes (centrilobular dark pigment on biopsy)
- All other liver function tests are normal
- Diagnosis:
- Urine coproporphyrin: total is normal, but isomer I constitutes > 80% of total (normally coproporphyrin III predominates at ~75%)
- Liver biopsy shows characteristic dark pigment
- Prognosis: Benign; no progressive liver damage
5. Rotor Syndrome
- Gene: Simultaneous mutations in both SLCO1B1 and SLCO1B3 (encoding OATP1B1 and OATP1B3) → impaired reuptake of bilirubin conjugates from the sinusoid back into hepatocytes (i.e., bilirubin conjugates that re-enter the circulation cannot be recaptured)
- Inheritance: Autosomal recessive
- Bilirubin: Conjugated hyperbilirubinemia, 3–7 mg/dL (occasionally up to 20 mg/dL); ~60% direct-reacting
- Clinical:
- Asymptomatic jaundice
- Liver biopsy is completely normal (no pigment — this distinguishes it from Dubin-Johnson)
- All other liver function tests are normal
- Diagnosis:
- Urine coproporphyrin: total is elevated 2–5 fold (vs. Dubin-Johnson where total is normal); coproporphyrin I < 80% of total
- Distinction from Dubin-Johnson: Rotor = elevated total coproporphyrin; Dubin-Johnson = normal total but abnormal ratio
FAMILIAL INTRAHEPATIC CHOLESTASIS SYNDROMES
6. Progressive Familial Intrahepatic Cholestasis (PFIC)
A group of autosomal recessive disorders causing inability to form and excrete bile, leading to chronic cholestasis and progressive liver disease.
| Type | Gene | Protein | GGT | Key Features |
|---|---|---|---|---|
| PFIC-1 | ATP8B1 | Aminophospholipid translocase (FIC1) | Normal/low | Presents in infancy; chronic cholestasis; progresses to cirrhosis; may recur after transplant |
| PFIC-2 | ABCB11 | Bile Salt Export Pump (BSEP) | Normal/low | Severe cholestasis from infancy; high risk of hepatocellular carcinoma (15%); requires liver transplant |
| PFIC-3 | ABCB4 | MDR3 (phosphatidylcholine flippase) | Elevated | Absent canalicular phospholipids → bile salt toxicity → ductular proliferation, portal fibrosis; responds to ursodeoxycholic acid (UDCA) |
- PFIC-1 and PFIC-2 are associated with low GGT (distinguishing from most other cholestatic diseases)
- PFIC-2 carries the highest risk of malignancy — surveillance is mandatory
- Benign Recurrent Intrahepatic Cholestasis (BRIC) shares mutations with PFIC-1 (ATP8B1) and PFIC-2 (ABCB11) but presents with intermittent reversible episodes rather than progressive disease
NEONATAL / PHYSIOLOGIC (Non-inherited but Developmental)
7. Neonatal Physiologic Jaundice
- UGT1A1 levels are low at birth, reaching adult levels only by 3–4 months of age
- Accelerated hemolysis of fetal hemoglobin further increases bilirubin load
- Transient unconjugated hyperbilirubinemia nearly universal in the first week of life
- Breastfeeding can exacerbate it (bilirubin-deconjugating enzymes in breast milk; progestational steroids inhibit UGT1A1)
- Treatment: Blue-light phototherapy (converts bilirubin to water-soluble photoisomers); phenobarbital if needed
- Kernicterus risk if unconjugated bilirubin > 20–25 mg/dL (exceeds albumin binding capacity)
Key Differentiating Points
| Feature | Crigler-Najjar I | Crigler-Najjar II | Gilbert | Dubin-Johnson | Rotor |
|---|---|---|---|---|---|
| Bilirubin type | Unconjugated | Unconjugated | Unconjugated | Conjugated | Conjugated |
| Bilirubin level | 18–45 mg/dL | 6–25 mg/dL | < 4 mg/dL | 2–5 mg/dL | 3–7 mg/dL |
| UGT1A1 | Absent | ≤10% | 10–33% | Normal | Normal |
| Liver histology | Normal | Normal | Normal | Black pigment | Normal |
| Phenobarbital | No response | Reduces bilirubin | Reduces bilirubin | N/A | N/A |
| Urine coproporphyrin | Normal | Normal | Normal | Total normal, I > 80% | Total elevated 2–5× |
| Prognosis | Fatal without Tx | Good | Excellent | Benign | Benign |
Sources: Robbins & Cotran Pathologic Basis of Disease; Harper's Illustrated Biochemistry 32nd Ed; Harrison's Principles of Internal Medicine 22nd Ed; Goldman-Cecil Medicine; Yamada's Textbook of Gastroenterology; The Harriet Lane Handbook 23rd Ed
Barter syndrome
Checking Date & Time
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
Bartter syndrome thick ascending limb ion transport NKCC2 ROMK
Reading File
Loading Image
Loading Image
Excellent — I have all the material needed. Here is a comprehensive answer.
Bartter Syndrome
Bartter syndrome is a genetically heterogeneous, autosomal recessive renal tubular disorder caused by loss-of-function mutations in transporters of the thick ascending limb (TAL) of the loop of Henle, resulting in impaired NaCl reabsorption, salt wasting, volume depletion, and secondary hyperreninemic hyperaldosteronism with hypokalemic metabolic alkalosis — all with normal to low blood pressure.
The clinical picture closely mimics chronic loop diuretic (furosemide) use.
Anatomy & Physiology of the TAL (Key to Understanding Bartter)
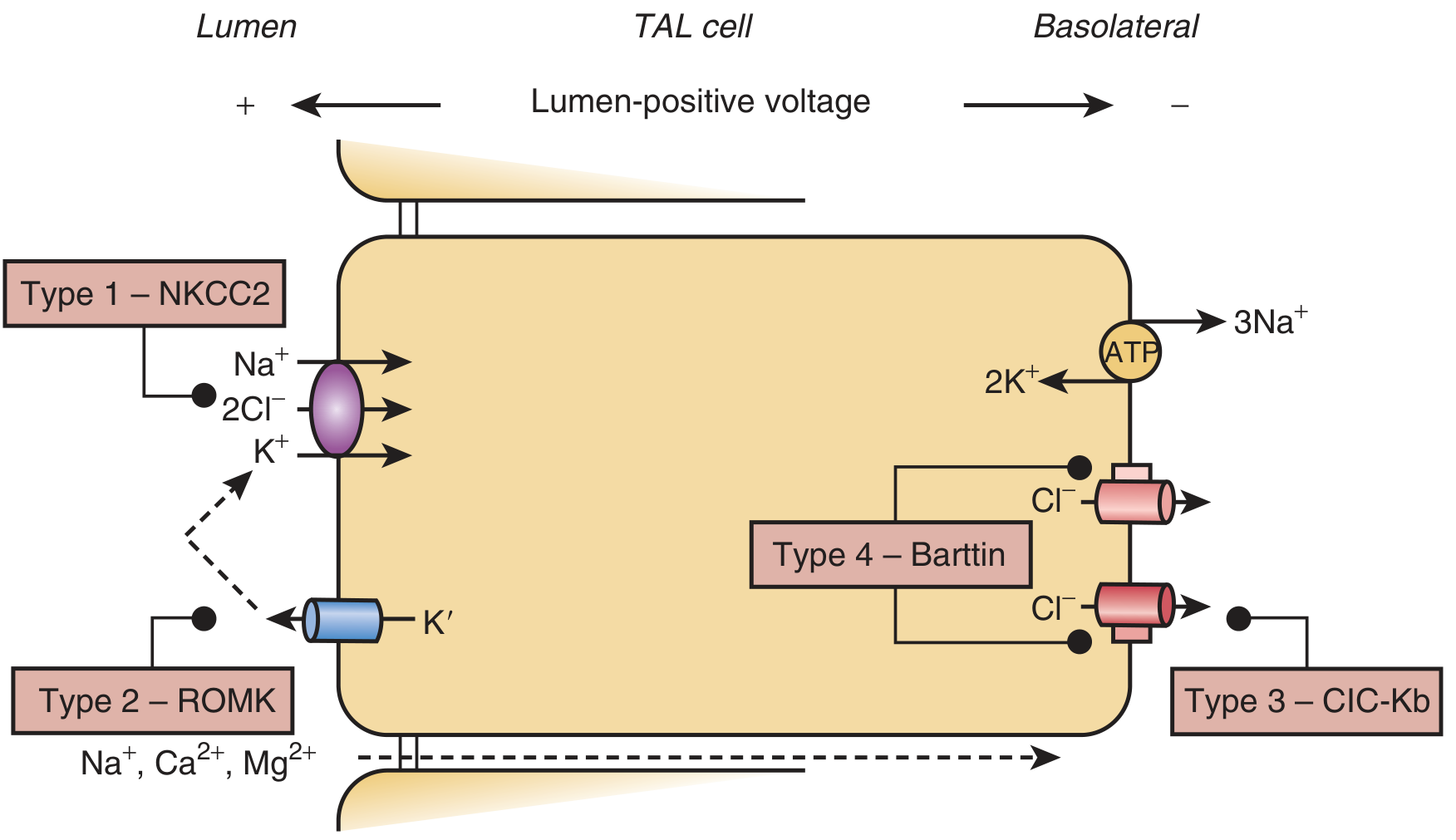
TAL cell showing transport systems disrupted in each Bartter type — Brenner & Rector's The Kidney
In the TAL:
- NKCC2 (apical) co-transports Na⁺, K⁺, and 2Cl⁻ into the cell
- ROMK (apical K⁺ channel) recycles K⁺ back into the lumen — this creates a lumen-positive transepithelial voltage
- This voltage drives paracellular Ca²⁺ and Mg²⁺ reabsorption
- Cl⁻ exits basolaterally via CLC-Kb chloride channels, which require the Barttin (BSND) subunit
- The Na⁺/K⁺-ATPase maintains the gradient
When any of these components fail → NaCl reabsorption collapses → salt wasting.
Genetic Classification (Types 1–5)
| Type | Gene | Protein | Phenotype | Key Feature |
|---|---|---|---|---|
| BS1 | SLC12A1 | NKCC2 | Antenatal | Polyhydramnios, hypercalciuria, nephrocalcinosis |
| BS2 | KCNJ1 | ROMK | Antenatal | Paradoxical neonatal hyperkalemia initially |
| BS3 | CLCNKB | CLC-Kb | Classic | Most variable; overlap with Gitelman; hypomagnesemia common |
| BS4 | BSND | Barttin | Antenatal + severe | Sensorineural deafness + progressive CKD |
| BS4b | CLCNKA + CLCNKB | CLC-Ka & CLC-Kb | Antenatal + severe | Both Cl⁻ channels lost → severe phenotype + deafness |
| BS5 | CASR | Calcium-sensing receptor (gain of function) | Variable | Activating mutation → inhibits ROMK → Bartter-like picture |
Inheritance: All types are autosomal recessive, except BS5 which involves an activating (gain-of-function) dominant mutation in CaSR.
Pathophysiology: The Chain of Events
Loss of NaCl reabsorption in TAL
↓
↓ NaCl delivery to macula densa
↓
Impaired tubuloglomerular feedback (TGF)
↓
↑ COX-2 activity in TAL & macula densa
↓
↑ Prostaglandin E₂ (PGE₂)
↓
↑ Renin → ↑ Angiotensin II → ↑ Aldosterone (hyperreninemic hyperaldosteronism)
↓ ↓
Afferent arteriolar dilation ↑ Na⁺ reabsorption in collecting duct
(vascular resistance to ANG II) ↑ K⁺ secretion → Hypokalemia
↑ H⁺ secretion → Metabolic alkalosis
Additionally:
- Juxtaglomerular apparatus hypertrophy (due to chronic renin hyperstimulation)
- Vascular unresponsiveness to angiotensin II → persistent normotension or hypotension despite high aldosterone
- Loss of lumen-positive voltage in TAL (Types 1 & 2) → hypercalciuria + nephrocalcinosis
Clinical Presentations
Antenatal / Neonatal Bartter (Types 1, 2, 4)
- Maternal polyhydramnios (fetal polyuria) → premature birth
- Postnatal: polyuria, polydipsia, vomiting, failure to thrive
- Hypercalciuria → nephrocalcinosis
- Marked electrolyte wasting (hypokalemia, hyponatremia, hypochloremia)
- Elevated plasma renin, aldosterone, and PGE₂
- Type 2 special feature: Neonates initially present with hyperkalemia and hyponatremia (mimicking pseudohypoaldosteronism type 1), because ROMK also mediates K⁺ secretion in the collecting duct; this resolves after a few weeks as alternative K⁺ channels compensate
Classic Bartter (Type 3 — CLCNKB)
- Onset: first decade of life (occasionally adolescence or adulthood)
- Symptoms: vomiting, polyuria, recurrent dehydration, muscle cramps, weakness
- Hypomagnesemia frequent (CLC-Kb expressed in both TAL and DCT)
- Can phenotypically overlap with Gitelman syndrome (some patients look like Gitelman)
- Electrolyte disturbances can be paradoxically more severe than antenatal forms
Bartter with Deafness (Type 4 — Barttin)
- Severe form of BS3 + sensorineural deafness (Barttin required for inner ear CLC-Ka/Kb function)
- Progressive CKD is common
- Barttin loss = functional loss of both CLC-Ka and CLC-Kb
Biochemical Profile
| Parameter | Finding |
|---|---|
| Serum K⁺ | ↓ (hypokalemia) |
| Serum Cl⁻ | ↓ (hypochloremia) |
| Blood pH / HCO₃⁻ | ↑ (metabolic alkalosis) |
| Serum Mg²⁺ | ↓ (esp. Type 3) or normal |
| Serum Ca²⁺ | Normal |
| Blood pressure | Normal or low (key distinguishing feature) |
| Plasma renin | ↑↑ |
| Plasma aldosterone | ↑↑ |
| Urinary Ca²⁺ | ↑ (Types 1, 2); normal or low in Type 3 |
| Urinary Cl⁻ | Normal or elevated (not low — distinguishes from vomiting) |
| Urinary K⁺ | ↑ (renal K⁺ wasting) |
| Urine PGE₂ | ↑ (antenatal forms) |
Differential Diagnosis
| Condition | BP | Urine Cl⁻ | Ca²⁺ excretion | Mg²⁺ | Notes |
|---|---|---|---|---|---|
| Bartter | Normal/Low | High | High | Normal/↓ | High renin/aldosterone |
| Gitelman | Normal/Low | High | Low (hypocalciuria) | ↓↓ | Thiazide-like, DCT defect |
| Diuretic abuse (loop) | Variable | High | High | Variable | Urine diuretic screen positive |
| Surreptitious vomiting | Normal/Low | Low | Normal | Normal | Low urine Cl⁻ is key |
| Primary hyperaldosteronism | High | High | Normal | Normal | Low renin |
| Liddle syndrome | High | High | Normal | Normal | Low renin, low aldosterone |
Key rule: Low blood pressure + high urine Cl⁻ + high renin + hypokalemic alkalosis = Bartter or Gitelman. Distinguish by urine Ca²⁺ and Mg²⁺.
Treatment
Acute (Neonates/Severe Episodes)
- IV saline for fluid and electrolyte correction
- Potassium chloride supplementation (oral or IV)
- Monitor closely — supplementation causes transient rise then urinary loss; smaller, more frequent doses are safer
Chronic / Maintenance
| Drug | Mechanism | Note |
|---|---|---|
| Indomethacin (NSAIDs) | COX inhibition → ↓ PGE₂ → ↓ renin → ↓ polyuria, improved growth | Mainstay for antenatal BS; risk of bowel perforation, NEC in neonates |
| Selective COX-2 inhibitors (celecoxib, rofecoxib) | More targeted PGE₂ inhibition | Proposed as safer alternative; cardiovascular risk in adults |
| Spironolactone / Eplerenone | Aldosterone antagonist → ↓ K⁺ wasting | Worsens salt wasting; combine with adequate salt |
| Amiloride | ENaC blocker → ↓ K⁺ secretion in CD | Useful esp. for Type 3 overlap with Gitelman |
| KCl supplements | Direct K⁺ replacement | Always necessary |
| Mg²⁺ supplements | Direct replacement | Especially Type 3/4 |
| ACE inhibitors | ↓ ANG II | Used in selected patients; risk of hypotension |
ACE inhibitors are generally not recommended in antenatal/severe forms due to hypotension risk.
Outcomes
- With appropriate therapy, most patients clinically improve significantly
- Progressive CKD is a concern, especially in Type 4 (Barttin mutations)
- Antenatal forms carry risk of extreme prematurity complications (intracranial hemorrhage, bronchopulmonary dysplasia)
- Severe hypokalemia can cause arrhythmias, paralysis, rhabdomyolysis, apnea
- Some patients diagnosed incidentally in adulthood with mild phenotypes
Sources: Brenner & Rector's The Kidney; Comprehensive Clinical Nephrology 7th Ed; National Kidney Foundation Primer on Kidney Diseases 8th Ed; Harrison's Principles of Internal Medicine 22nd Ed
This is a shared conversation. Sign in to Orris to start your own chat.