Osteoarthritis aetiopathogenesis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"osteoarthritis"[MeSH Terms] AND "pathogenesis"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Osteoarthritis: Aetiopathogenesis
Definition
Osteoarthritis (OA) is characterized by failed repair of synovial joint tissues after intra-articular stress, resulting in breakdown of cartilage and bone with symptoms of pain, stiffness, and functional disability. Despite the suffix "-itis," OA is primarily a degenerative disorder of articular cartilage; inflammation acts as a secondary but important contributor. - Goldman-Cecil Medicine, p. 2763; Robbins & Kumar Basic Pathology, p. 789
Classification
| Type | Definition | Cause |
|---|---|---|
| Primary (idiopathic) | Appears insidiously as an aging phenomenon | No identifiable predisposing condition; oligoarticular |
| Secondary | Appears in younger individuals (~5% of cases) | Joint deformity, prior trauma, systemic disease (diabetes, obesity) |
Prevalence increases exponentially beyond age 50; approximately 40% of people over 70 are affected. - Robbins, p. 789
Aetiology and Risk Factors
1. Biomechanical Factors
The core initiating event is an imbalance between the mechanical load on articular cartilage and its capacity to bear that load.
- Overloaded or incorrectly loaded joints (post-traumatic deformities, joint dysplasia, axis deviations, instabilities)
- Sports injuries
- Obesity - Mendelian randomization studies confirm a causative role of BMI in OA
- Weight-bearing joints (hips, knees, lumbar/cervical spine) are preferentially affected
2. Genetic Factors
Genome-wide association studies (GWAS) with >800,000 samples have identified more than 80 OA risk loci, confirming that OA has a significant polygenic component. Key genes include:
- GDF5 (chromosome 20): codes for a growth factor important in chondrogenesis and bone growth; risk allele is inversely associated with height
- RUNX2, SMAD3, PTHLH: important in skeletal and bone development - highlighting bone morphology's contribution
- DOT1L: another height-inversely-associated locus
- Genes affecting the immune/inflammatory response, skeletal development, and joint degeneration are all represented across loci
Different genetic variants associate with different joint sites (e.g., hip vs. knee OA). - Firestein & Kelley's Rheumatology, p. 499
3. Other Risk Factors
- Age (cartilage is bradytrophic - avascular and barely regenerates)
- Female sex
- Lack of exercise, poor nutrition
- Prior inflammatory joint disease
Pathogenesis: Step-by-Step
The following diagram from Robbins & Kumar summarizes the sequence:
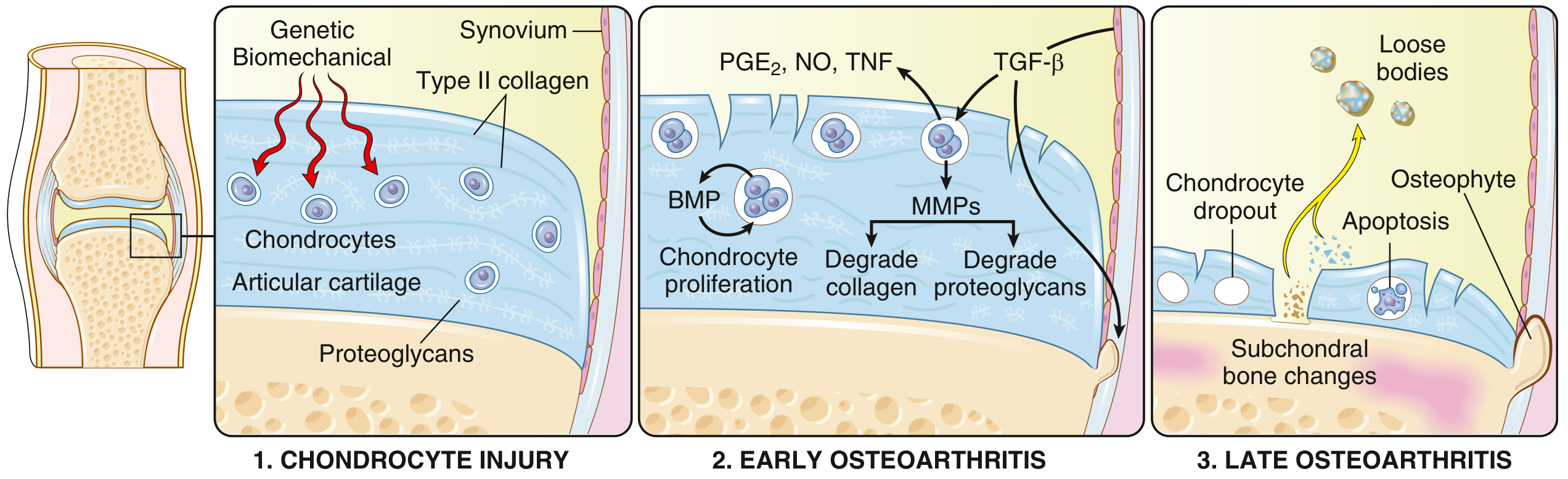
FIG: OA is initiated by chondrocyte injury in a genetically predisposed patient (1), leading to matrix changes; degradation exceeds repair in early OA (2); late OA shows loss of matrix and chondrocytes with subchondral bone damage (3). - Robbins & Kumar Basic Pathology, p. 790
Stage 1 - Chondrocyte Injury (Initiation)
Biomechanical stress disrupts the normal flow of interstitial fluid into avascular articular cartilage. Because cartilage has no blood vessels or perichondrium, it has severely limited regenerative capacity. Initial stress injures chondrocytes and disrupts the type II collagen network.
Stage 2 - Early OA: Degradation vs. Failed Repair
- Injured chondrocytes proliferate (forming clusters visible histologically) in a failed repair response
- They secrete matrix metalloproteinases (MMPs), particularly MMP-13, which cleave the type II collagen fibers
- ADAMTS5 (aggrecanase) degrades proteoglycans (aggrecan)
Key regulatory mechanism disrupted in OA: In normal cartilage, MMP-13 and ADAMTS5 are constitutively produced but kept in check by LRP1-mediated endocytic clearance. In OA:
- Chondrocytes overproduce MT1-MMP and ADAM17
- These enzymes shed the ectodomain of LRP1
- Without LRP1, MMP-13 and ADAMTS5 accumulate and drive matrix destruction
- Firestein & Kelley's Rheumatology, p. 183
Consequences of matrix loss:
- Water content of the matrix increases
- Proteoglycan concentration decreases
- Horizontal type II collagen fibers are cleaved, yielding surface fissures and clefts (fibrillation)
- Cartilage becomes granular and soft
Cytokine/mediator amplification loop:
Chondrocytes, synovial cells, and recruited macrophages release:
| Mediator | Effect |
|---|---|
| IL-1, TNF | Induce MMP expression; directly promote cartilage catabolism |
| IL-6 | Amplifies inflammatory signaling |
| TGF-β | Paradoxically induces MMP production; also promotes osteophyte formation via BMP pathway |
| PGE2 (prostaglandins) | Pain, inflammation |
| Nitric oxide (NO) | Promotes chondrocyte apoptosis |
| BMPs (Bone Morphogenetic Proteins) | Contribute to osteophyte formation at joint margins |
Stage 3 - Late OA: Structural Failure
- Chondrocyte dropout by apoptosis
- Full-thickness sloughing of cartilage segments
- Dislodged fragments form loose bodies ("joint mice") in the joint space
- Exposed subchondral bone is polished by friction = eburnation (ivory-like appearance)
- Microfractures in subchondral bone allow synovial fluid to enter via a ball-valve mechanism = subchondral cysts (geodes)
- TGF-β/BMP signaling drives peripheral osteophyte formation (fibrocartilage-capped bony outgrowths that gradually ossify)
- Synovium shows mild congestion, fibrosis, and scattered chronic inflammatory cells
Histopathology
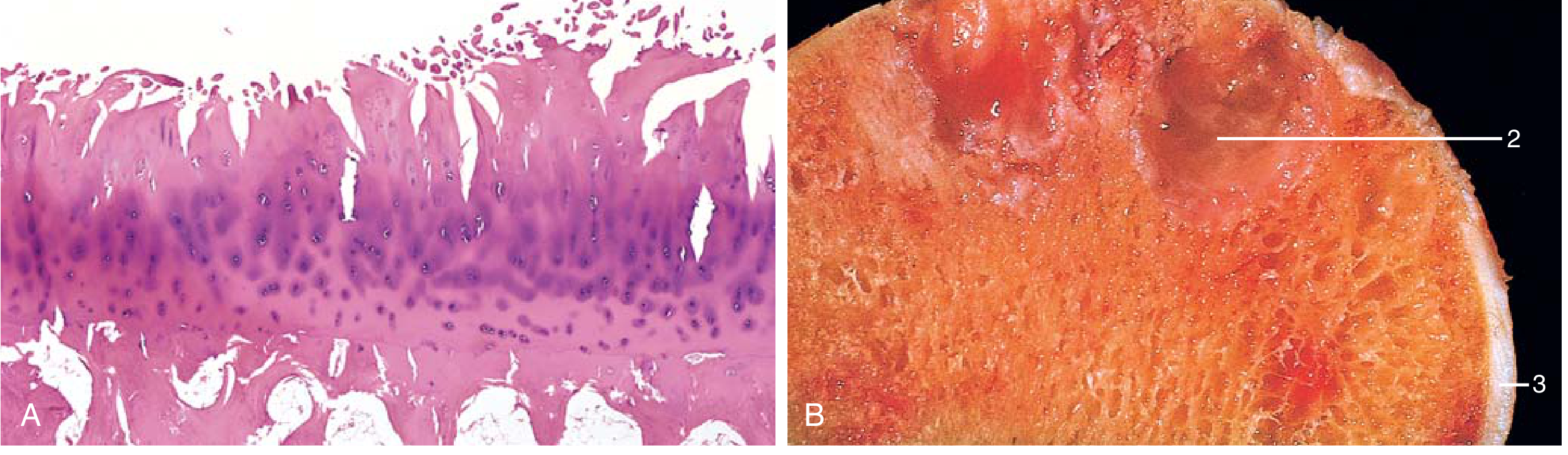
FIG 19.32: (A) Fibrillation of articular cartilage on H&E. (B) Eburnated articular surface (ivory-like exposed bone), with subchondral cyst (2) and residual cartilage (3). - Robbins & Kumar, p. 791
In early OA, Safranin-O staining (which stains proteoglycans) shows loss of red staining, indicating proteoglycan depletion. Quiescent chondrocytes become activated and form clusters. The tidemark (interface between articular and calcified cartilage) advances and may duplicate, and vascular penetration from subchondral bone occurs. - Firestein & Kelley's Rheumatology, p. 75
The Whole-Joint Disease Concept
Modern thinking views OA as a disease of the whole joint organ, not just cartilage:
| Structure | Changes in OA |
|---|---|
| Articular cartilage | Fibrillation, proteoglycan loss, full-thickness erosion |
| Subchondral bone | Sclerosis, bone marrow lesions (BMLs), cyst formation, eburnation |
| Synovium | Low-grade synovitis, inflammatory cell infiltration (secondary) |
| Osteophytes | Peripheral bony outgrowths (TGF-β/BMP driven) |
| Ligaments & periarticular muscles | Laxity, atrophy |
| Joint capsule | Fibrosis |
Pain Mechanisms
OA pain is multifactorial - the poor correlation between structural severity and symptom severity reflects this complexity:
- Synovitis - inflammatory mediators sensitize synovial nociceptors; NSAIDs and intra-articular corticosteroids are effective analgesics in OA
- Bone marrow lesions (BMLs) - associated with inflammatory cell infiltration, cytokine and NGF expression, and sensory nerve terminal ingrowth into subchondral bone
- Biomechanical - weight-bearing pain; exercises, braces, osteotomy, and arthroplasty all reduce pain by altering mechanics
- NGF/peripheral sensitization - NGF-blocking antibodies (e.g., tanezumab) effectively reduce OA pain, highlighting peripheral nociceptor sensitization as a key driver
- Central sensitization - quantitative sensory testing (QST) demonstrates widespread hyperalgesia; fibromyalgia comorbidity is common. After successful arthroplasty, central sensitization may resolve, suggesting the joint itself drives central pain facilitation
- Firestein & Kelley's Rheumatology, pp. 4585-4660
Summary: Key Pathogenic Cascade
Biomechanical stress + Genetic predisposition
↓
Chondrocyte injury
↓
↑ MMPs (MMP-13) + ↑ ADAMTS5 (LRP1 regulation lost)
↓
Collagen II + Proteoglycan degradation
↓
IL-1, TNF, PGE2, NO, TGF-β, BMP release
↓
Amplification loop → more matrix destruction
↓
Chondrocyte apoptosis → cartilage loss
↓
Subchondral bone exposure → eburnation, cysts, BMLs
↓
Osteophyte formation at joint margins
↓
Synovitis (secondary) + peripheral/central sensitization
↓
Pain, stiffness, functional disability
Sources:
- Robbins & Kumar Basic Pathology, pp. 789-791
- Firestein & Kelley's Textbook of Rheumatology, pp. 75, 183, 499, 4585-4676
- Goldman-Cecil Medicine, p. 2763
- General Anatomy and Musculoskeletal System (THIEME Atlas), pp. 1895-1905
This is a shared conversation. Sign in to Orris to start your own chat.